Department of Pediatric Neurology, Catholic University, Rome, Italy.
Centro Clinico Nemo, Fondazione Policlinico Agostino Gemelli IRCCS, Rome, Italy.
J Neurol. 2023 Aug;270(8):3896-3913. doi: 10.1007/s00415-023-11687-1. Epub 2023 Apr 28.
Strategic Targeting of Registries and International Database of Excellence (STRIDE) is an ongoing, international, multicenter registry of real-world ataluren use in individuals with nonsense mutation Duchenne muscular dystrophy (nmDMD) in clinical practice. This updated interim report (data cut-off: January 31, 2022), describes STRIDE patient characteristics and ataluren safety data, as well as the effectiveness of ataluren plus standard of care (SoC) in STRIDE versus SoC alone in the Cooperative International Neuromuscular Research Group (CINRG) Duchenne Natural History Study (DNHS).
Patients are followed up from enrollment for at least 5 years or until study withdrawal. Propensity score matching was performed to identify STRIDE and CINRG DNHS patients who were comparable in established predictors of disease progression.
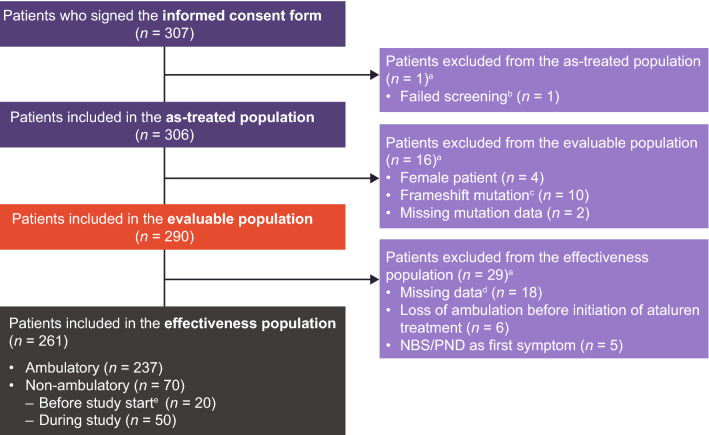
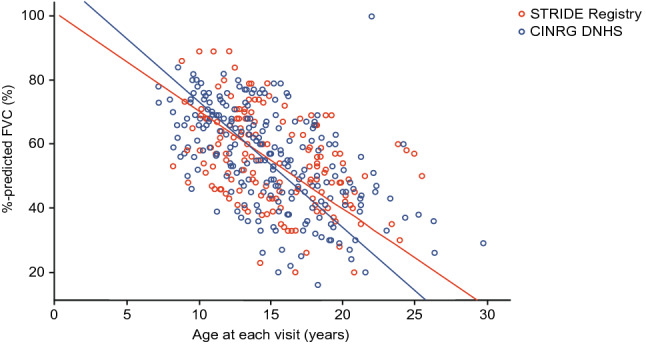
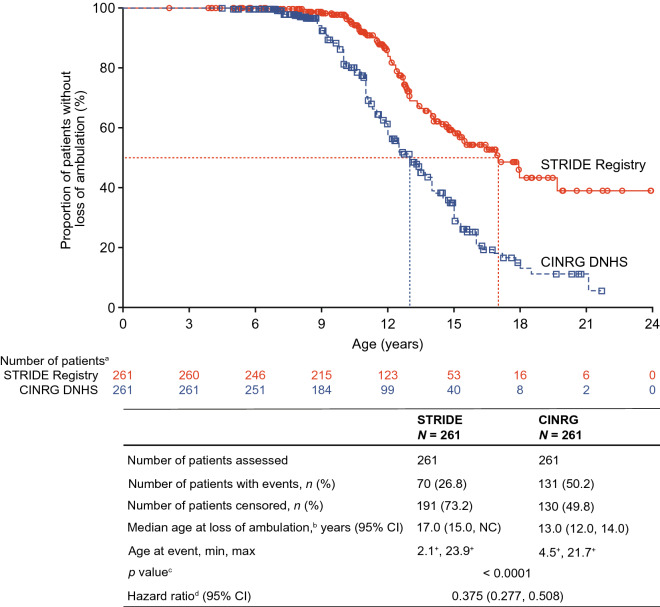
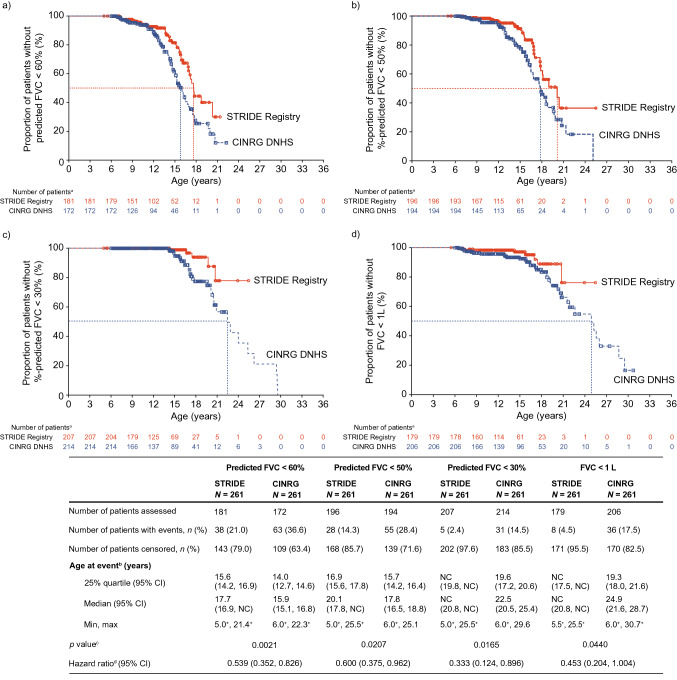
As of January 31, 2022, 307 patients were enrolled from 14 countries. Mean (standard deviation [SD]) ages at first symptoms and at genetic diagnosis were 2.9 (1.7) years and 4.5 (3.7) years, respectively. Mean (SD) duration of ataluren exposure was 1671 (56.8) days. Ataluren had a favorable safety profile; most treatment-emergent adverse events were mild or moderate and unrelated to ataluren. Kaplan-Meier analyses demonstrated that ataluren plus SoC significantly delayed age at loss of ambulation by 4 years (p < 0.0001) and age at decline to %-predicted forced vital capacity of < 60% and < 50% by 1.8 years (p = 0.0021) and 2.3 years (p = 0.0207), respectively, compared with SoC alone.
Long-term, real-world treatment with ataluren plus SoC delays several disease progression milestones in individuals with nmDMD. NCT02369731; registration date: February 24, 2015.
战略目标登记册和国际卓越数据库(STRIDE)是一项正在进行的、国际性的、多中心登记册,用于记录在临床实践中使用非义突变型杜氏肌营养不良症(nmDMD)的个体的阿托伦的真实世界使用情况。本更新的中期报告(数据截止日期:2022 年 1 月 31 日)描述了 STRIDE 患者的特征和阿托伦的安全性数据,以及阿托伦联合标准治疗(SoC)与单独 SoC 在合作国际神经肌肉研究组(CINRG)杜氏自然史研究(DNHS)中的疗效。
患者从入组开始至少随访 5 年或直至研究退出。采用倾向评分匹配来识别 STRIDE 和 CINRG DNHS 患者,这些患者在疾病进展的既定预测因素方面具有可比性。
截至 2022 年 1 月 31 日,来自 14 个国家的 307 名患者入组。首次症状和基因诊断的平均(标准差 [SD])年龄分别为 2.9(1.7)岁和 4.5(3.7)岁。阿托伦暴露的平均(SD)持续时间为 1671(56.8)天。阿托伦具有良好的安全性特征;大多数治疗出现的不良事件为轻度或中度,与阿托伦无关。Kaplan-Meier 分析表明,阿托伦联合 SoC 显著将丧失步行能力的年龄推迟了 4 岁(p<0.0001),以及下降至预测用力肺活量的<60%和<50%的年龄分别推迟了 1.8 岁(p=0.0021)和 2.3 岁(p=0.0207),与单独 SoC 相比。
长期、真实世界的阿托伦联合 SoC 治疗可延缓 nmDMD 患者的多个疾病进展里程碑。NCT02369731;注册日期:2015 年 2 月 24 日。