Zeng Jianfeng, Lai Cong, Luo Jianwei, Li Li
Department of Anesthesiology, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou, China.
Department of Urology, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou, China.
Front Neurosci. 2023 Apr 14;17:1134330. doi: 10.3389/fnins.2023.1134330. eCollection 2023.
Neuropathic pain as a complex chronic disease that occurs after neurological injury, however the underlying mechanisms are not clarified in detail, hence therapeutic options are limited. The purpose of this study was to explore potential hub genes for neuropathic pain and evaluate the clinical application of these genes in predicting neuropathic pain.
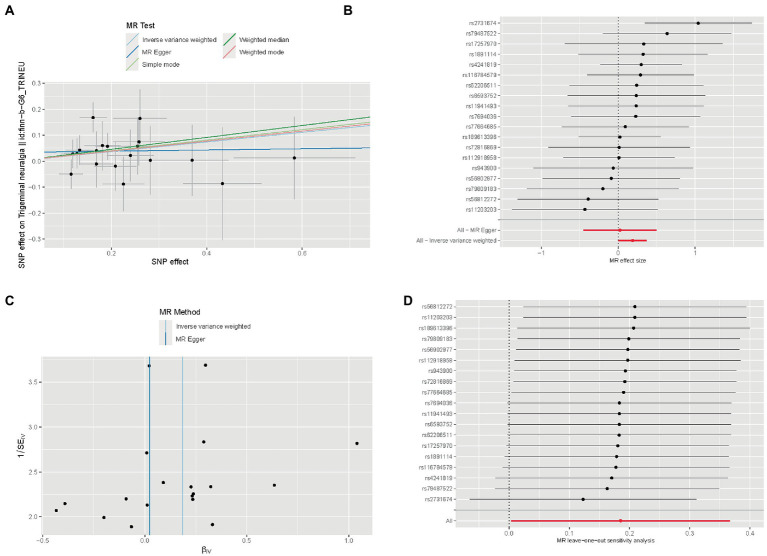
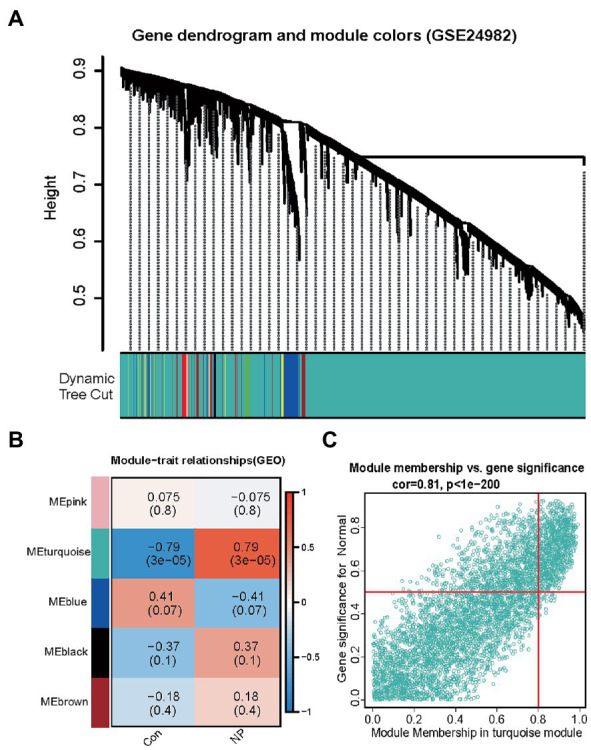
Differentially expressed analysis and weighted gene co-expression network analysis (WGCNA) was used to explore new neuropathic pain susceptibility modules and hub genes. KEGG and GO analyses was utilized to explore the potential role of these hub genes. Nomogram model and ROC curves was established to evaluate the diagnostic efficacy of hub genes. Additionally, the correlation of IL-2 with immune infiltration was explored. Finally, a Mendelian randomization study was conducted to determine the causal effect of IL-2 on neuropathic pain based on genome-wide association studies.
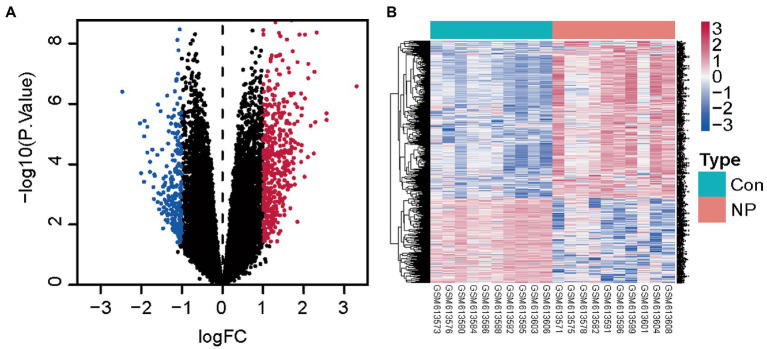
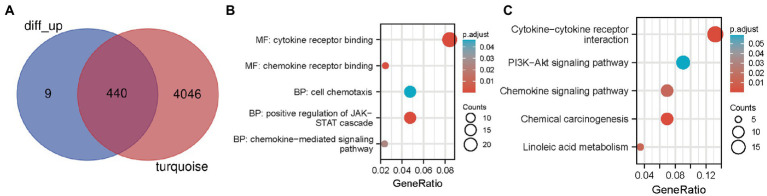
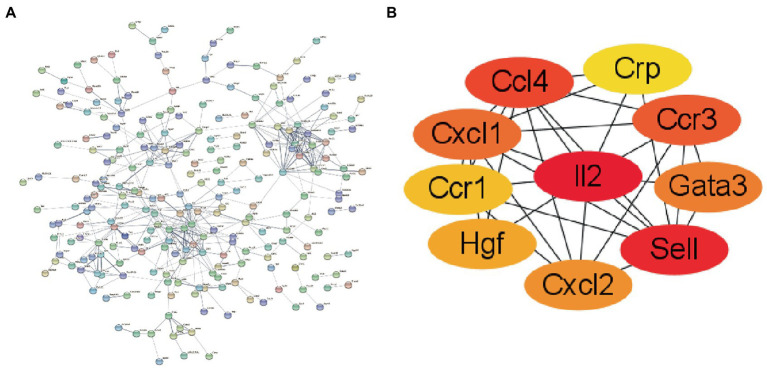
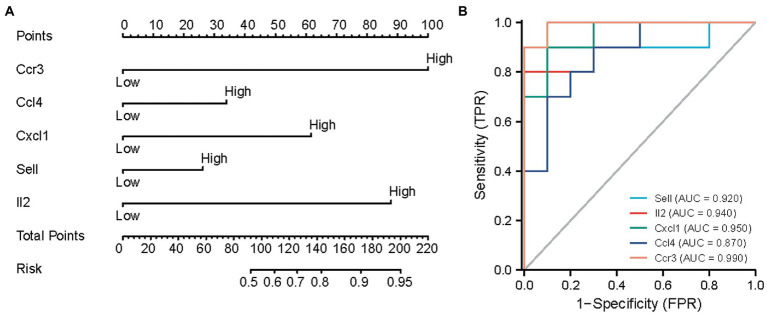
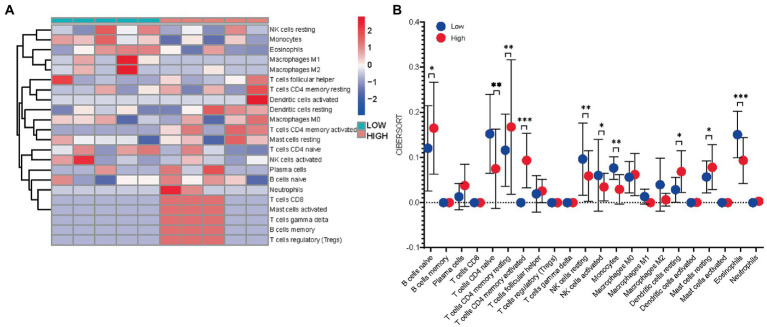
WGCNA was performed to establish the networks of gene co-expression, screen for the most relevant module, and screen for 440 overlapping WGCNA-derived key genes. GO and KEGG pathway enrichment analyses demonstrated that the key genes were correlated with cytokine receptor binding, chemokine receptor binding, positive regulation of JAK-STAT cascade, chemokine-mediated signaling pathway, PI3K-AKT pathway and chemokine pathway. Through Cytoscape software, top ten up-regulated genes with high scores were IL2, SMELL, CCL4, CCR3, CXCL1, CCR1, HGF, CXCL2, GATA3, and CRP. In addition, nomogram model performed well in predicting neuropathic pain risk, and with the ROC curve, the model was showed to be effective in diagnosis. Finally, IL2 was selected and we observed that IL2 was causally associated with immune cell infiltrates in trigeminal neuralgia. In inverse variance weighting, we found that IL2 was associated with the risk of trigeminal neuralgia with an OR of 1.203 (95% CI = 1.004-1.443, = 0.045).
We constructed a WGCNA-based co-expression network and identified neuropathic pain-related hub genes, which may offer further insight into pre-symptomatic diagnostic approaches and may be useful for the study of molecular mechanisms for understanding neuropathic pain risk genes.
神经性疼痛是一种在神经损伤后发生的复杂慢性疾病,但其潜在机制尚未完全阐明,因此治疗选择有限。本研究旨在探索神经性疼痛的潜在关键基因,并评估这些基因在预测神经性疼痛中的临床应用。
采用差异表达分析和加权基因共表达网络分析(WGCNA)来探索新的神经性疼痛易感性模块和关键基因。利用KEGG和GO分析来探索这些关键基因的潜在作用。建立列线图模型和ROC曲线以评估关键基因的诊断效能。此外,还探讨了IL-2与免疫浸润的相关性。最后,基于全基因组关联研究进行孟德尔随机化研究,以确定IL-2对神经性疼痛的因果效应。
进行WGCNA以建立基因共表达网络,筛选最相关的模块,并筛选出440个重叠的WGCNA衍生关键基因。GO和KEGG通路富集分析表明,关键基因与细胞因子受体结合、趋化因子受体结合、JAK-STAT级联的正调控、趋化因子介导的信号通路、PI3K-AKT通路和趋化因子通路相关。通过Cytoscape软件,得分最高的前十个上调基因是IL2、SMELL、CCL4、CCR3、CXCL1、CCR1、HGF、CXCL2、GATA3和CRP。此外,列线图模型在预测神经性疼痛风险方面表现良好,通过ROC曲线显示该模型在诊断中有效。最后,选择IL2,我们观察到IL2与三叉神经痛中的免疫细胞浸润存在因果关系。在逆方差加权中,我们发现IL2与三叉神经痛风险相关,OR为1.203(95%CI = 1.004 - 1.443,P = 0.045)。
我们构建了基于WGCNA的共表达网络并鉴定出神经性疼痛相关的关键基因,这可能为症状前诊断方法提供进一步的见解,并可能有助于研究理解神经性疼痛风险基因的分子机制。