Feng Zhi-Wei, Tang Yu-Chen, Sheng Xiao-Yun, Wang Sheng-Hong, Wang Yao-Bin, Liu Zhong-Cheng, Liu Jin-Min, Geng Bin, Xia Ya-Yi
Department of Orthopaedics, Lanzhou University Second Hospital, Lanzhou, China.
Gansu Province Orthopaedic Clinical Medicine Research Center, Lanzhou, China.
Heliyon. 2023 Jan 10;9(1):e12799. doi: 10.1016/j.heliyon.2023.e12799. eCollection 2023 Jan.
Rheumatoid arthritis (RA) is an autoimmune disease that affects individuals of all ages. The basic pathological manifestations are synovial inflammation, pannus formation, and erosion of articular cartilage, bone destruction will eventually lead to joint deformities and loss of function. However, the specific molecular mechanisms of synovitis tissue in RA are still unclear. Therefore, this study aimed to screen and explore the potential hub genes and immune cell infiltration in RA.
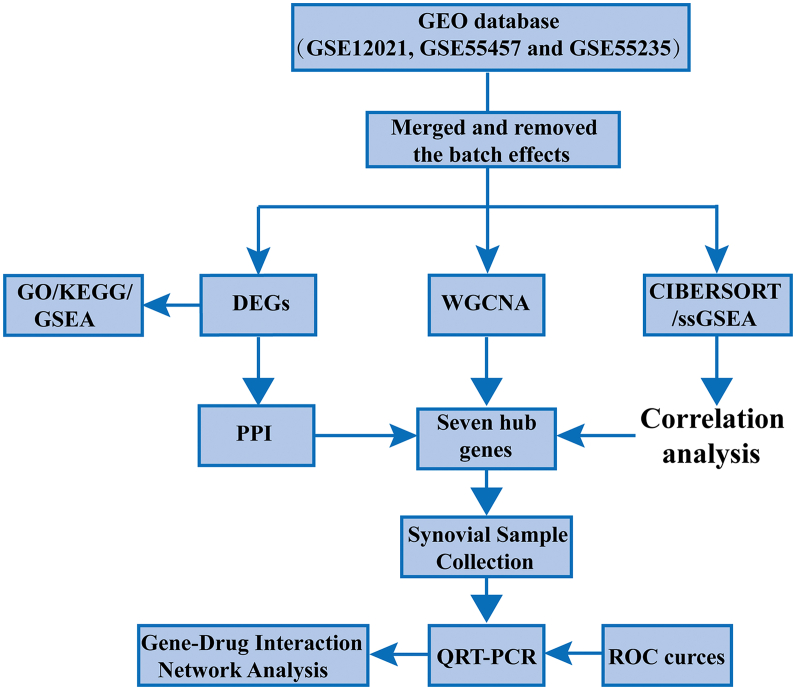
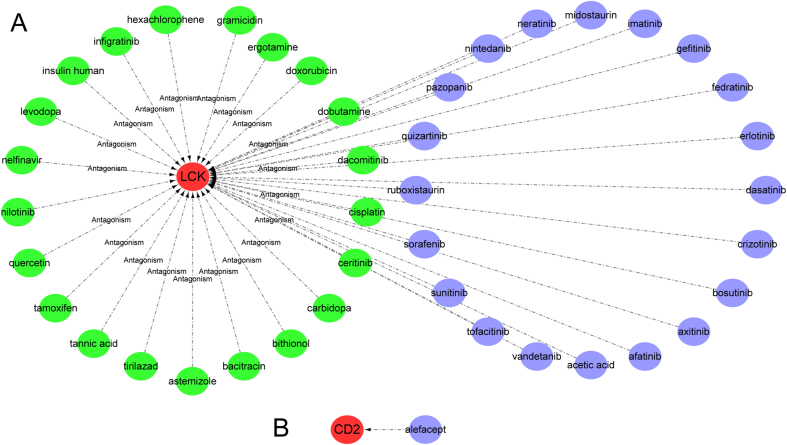
Three microarray datasets (GSE12021, GSE55457, and GSE55235), from the Gene Expression Omnibus (GEO) database, have been analyzed to explore the potential hub genes and immune cell infiltration in RA. First, the LIMMA package was used to screen the differentially expression genes (DEGs) after removing the batch effect. Then the clusterProfiler package was used to perform functional enrichment analyses. Second, through weighted coexpression network analysis (WGCNA), the key module was identified in the coexpression network of the gene set. Third, the protein-protein interaction (PPI) network was constructed through STRING website and the module analysis was performed using Cytoscape software. Fourth, the CIBERSORT and ssGSEA algorithm were used to analyze the immune status of RA and healthy synovial tissue, and the associations between immune cell infiltration and RA-related diagnostic biomarkers were evaluated. Fifth, we used the quantitative reverse transcription-polymerase chain reaction (qRT-PCR) to validate the expression levels of the hub genes, and ROC curve analysis of hub genes for discriminating between RA and healthy tissue. Finally, the gene-drug interaction network was constructed using DrugCentral database, and identification of drug molecules based on hub genes using the Drug Signature Database (DSigDB) by Enrichr.
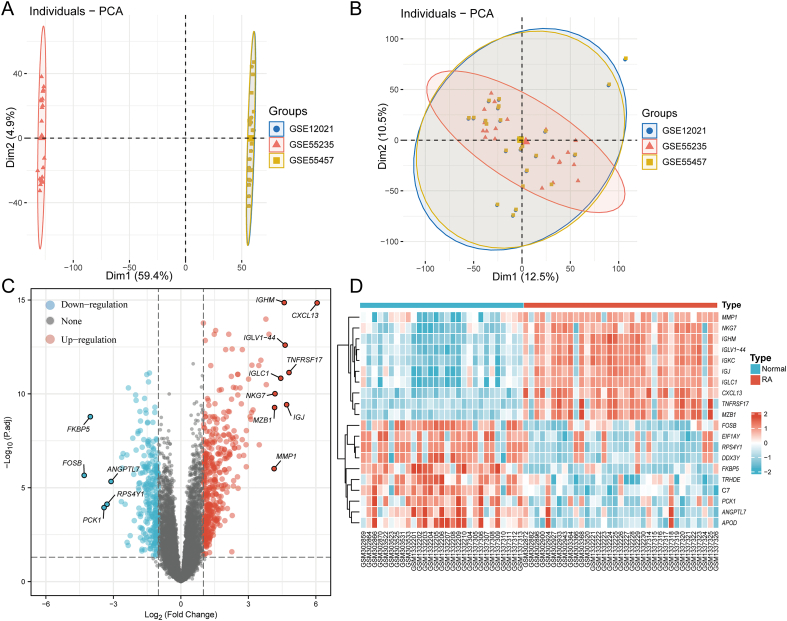
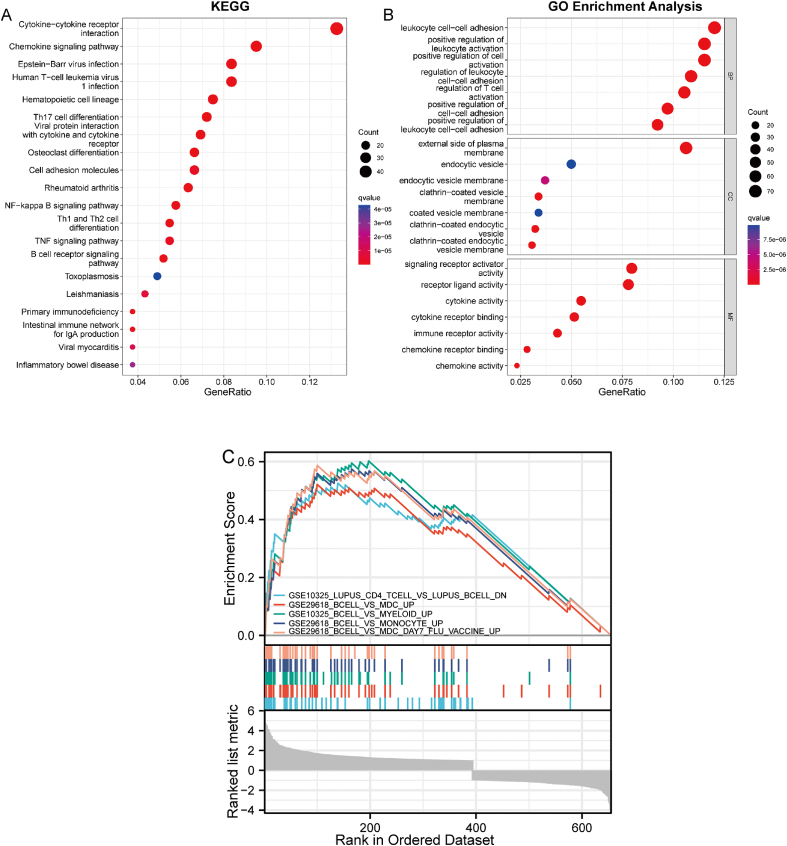
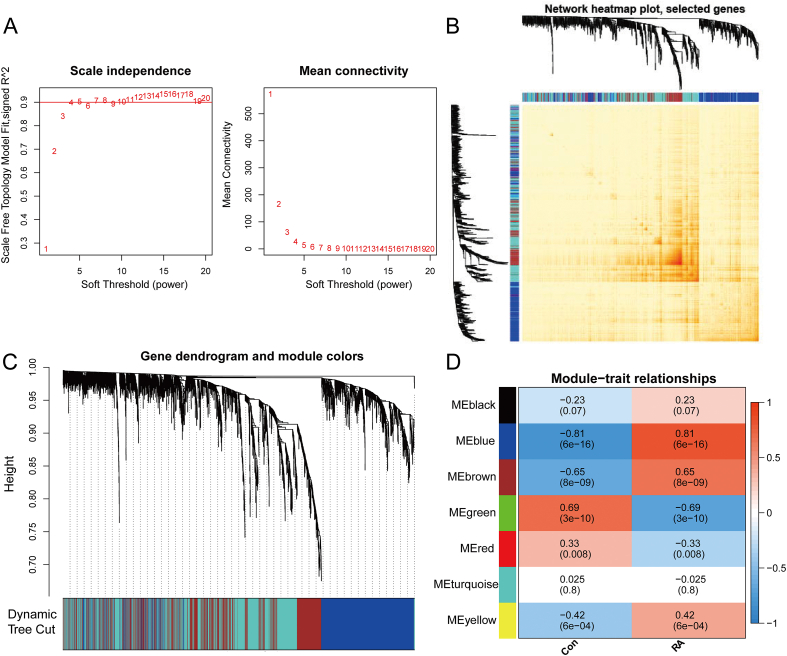
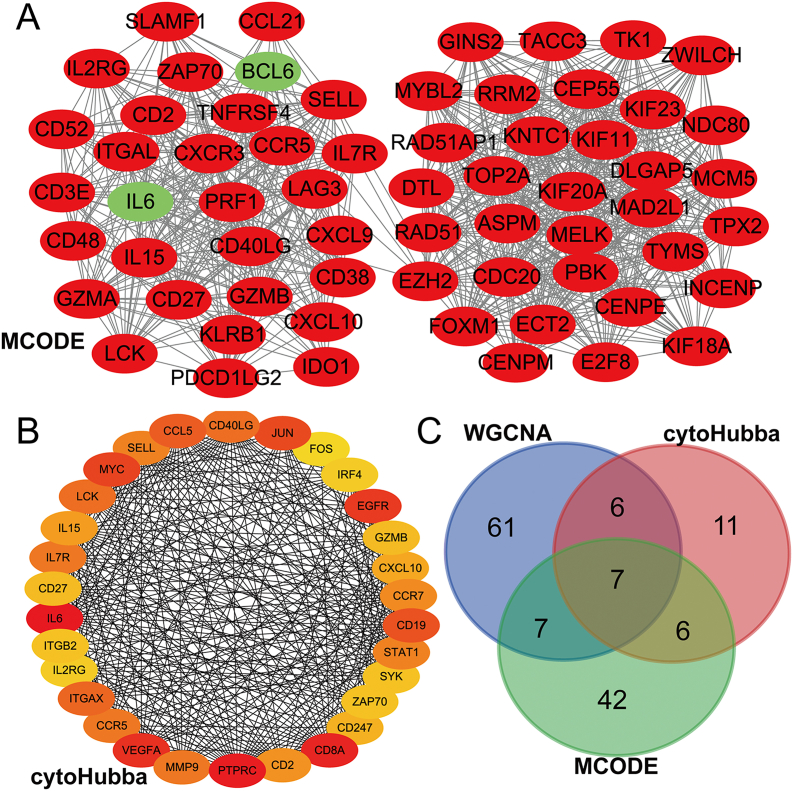
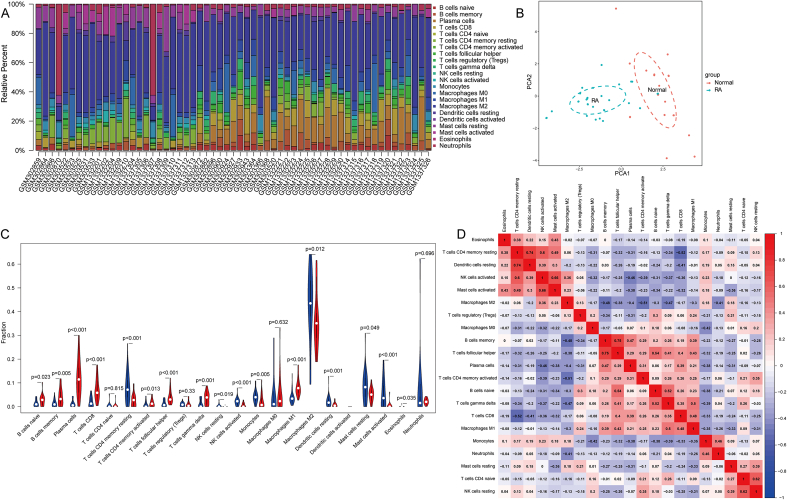
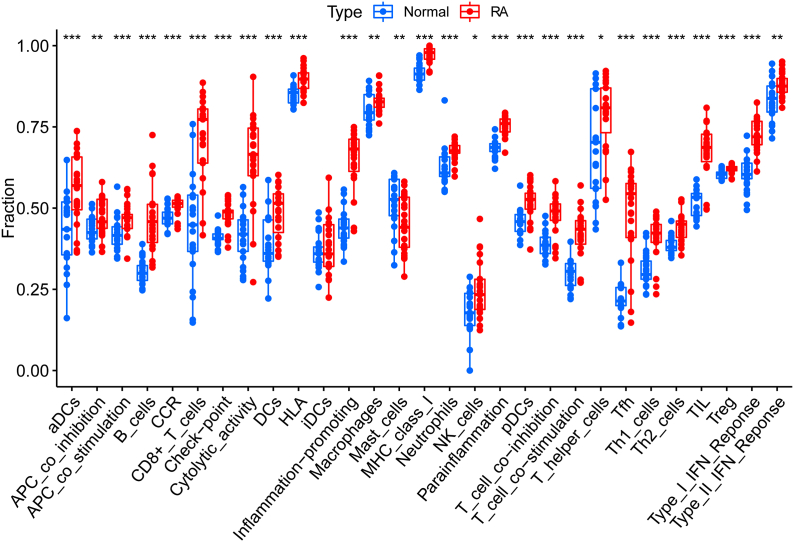
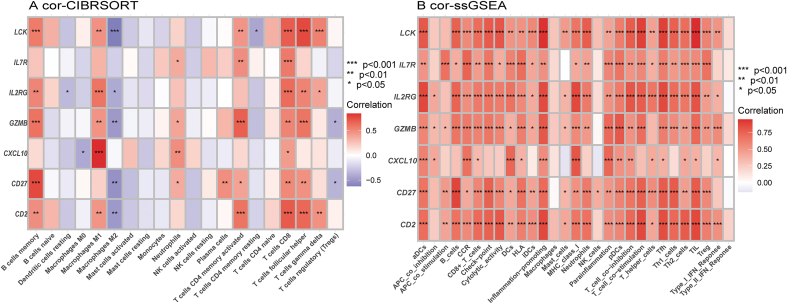
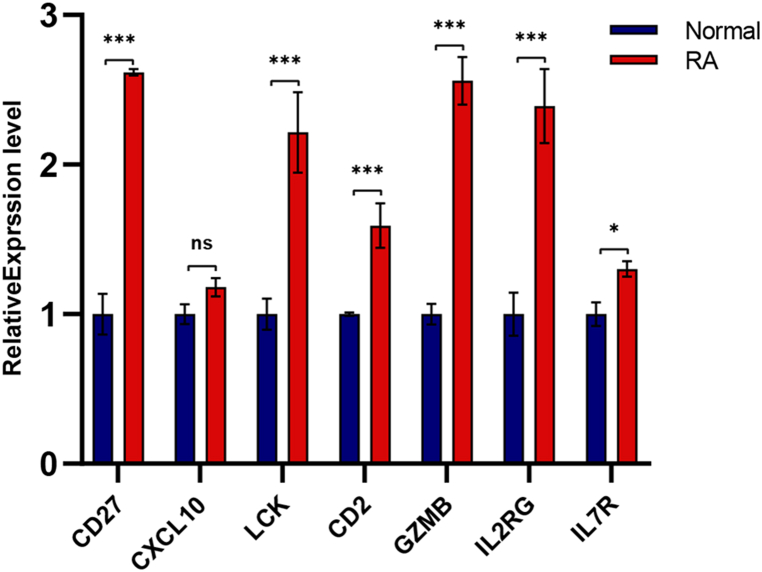
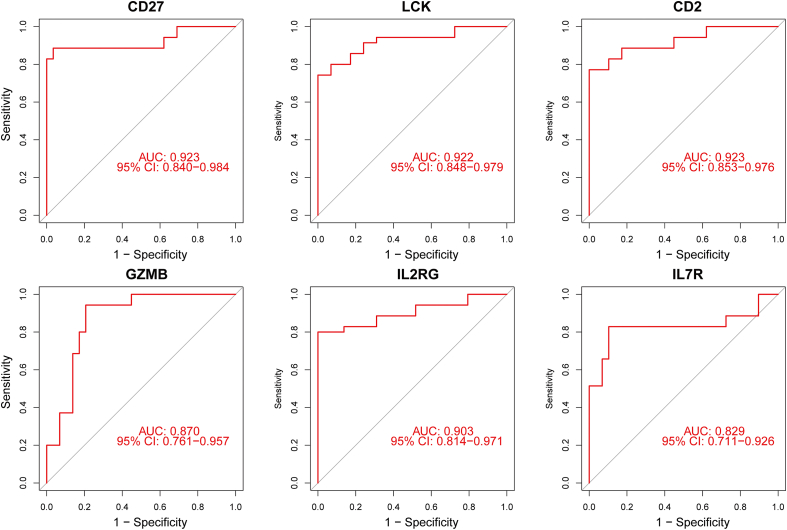
A total of 679 DEGs were identified, containing 270 downregulated genes and 409 upregulated genes. DEGs were primarily enriched in immune response and chemokine signaling pathways, according to functional enrichment analysis of DEGs. WGCNA explored the co-expression network of the gene set and identified key modules, the blue module was selected as the key module associated with RA. Seven hub genes are identified when PPI network and WGCNA core modules are intersected. Immune infiltration analysis using CIBERSORT and ssGSEA algorithms revealed that multiple types of immune infiltration were found to be upregulated in RA tissue compared to normal tissue. Furthermore, the levels of 7 hub genes were closely related to the relative proportions of multiple immune cells in RA. The results of the qRT-PCR demonstrated that the relative expression levels of 6 hub genes (CD27, LCK, CD2, GZMB, IL7R, and IL2RG) were up-regulated in RA synovial tissue, compared with normal tissue. Simultaneously, ROC curves indicated that the above 6 hub genes had strong biomarker potential for RA (AUC >0.8).
Through bioinformatics analysis and qRT-PCR experiment, our study ultimately discovered 6 hub genes (CD27, LCK, CD2, GZMB, IL7R, and IL2RG) that closely related to RA. These findings may provide valuable direction for future RA clinical diagnosis, treatment, and associated research.
类风湿关节炎(RA)是一种影响各年龄段人群的自身免疫性疾病。其基本病理表现为滑膜炎、血管翳形成以及关节软骨侵蚀,最终会导致骨质破坏、关节畸形和功能丧失。然而,RA中滑膜组织的具体分子机制仍不清楚。因此,本研究旨在筛选并探索RA中的潜在核心基因和免疫细胞浸润情况。
对来自基因表达综合数据库(GEO)的三个微阵列数据集(GSE12021、GSE55457和GSE55235)进行分析,以探索RA中的潜在核心基因和免疫细胞浸润情况。首先,使用LIMMA软件包在去除批次效应后筛选差异表达基因(DEGs)。然后使用clusterProfiler软件包进行功能富集分析。其次,通过加权共表达网络分析(WGCNA)在基因集的共表达网络中识别关键模块。第三,通过STRING网站构建蛋白质-蛋白质相互作用(PPI)网络,并使用Cytoscape软件进行模块分析。第四,使用CIBERSORT和ssGSEA算法分析RA和健康滑膜组织的免疫状态,并评估免疫细胞浸润与RA相关诊断生物标志物之间的关联。第五,我们使用定量逆转录-聚合酶链反应(qRT-PCR)验证核心基因的表达水平,并对核心基因进行ROC曲线分析以区分RA和健康组织。最后,使用DrugCentral数据库构建基因-药物相互作用网络,并通过Enrichr使用药物特征数据库(DSigDB)基于核心基因鉴定药物分子。
共鉴定出679个DEGs,其中包括270个下调基因和409个上调基因。根据DEGs的功能富集分析,DEGs主要富集于免疫应答和趋化因子信号通路。WGCNA探索了基因集的共表达网络并识别出关键模块,蓝色模块被选为与RA相关的关键模块。当PPI网络和WGCNA核心模块相交时,鉴定出7个核心基因。使用CIBERSORT和ssGSEA算法进行免疫浸润分析发现,与正常组织相比,RA组织中多种类型的免疫浸润上调。此外,7个核心基因的水平与RA中多种免疫细胞的相对比例密切相关。qRT-PCR结果表明,与正常组织相比,6个核心基因(CD27、LCK、CD2、GZMB、IL7R和IL2RG)在RA滑膜组织中的相对表达水平上调。同时,ROC曲线表明上述6个核心基因对RA具有较强的生物标志物潜力(AUC>0.8)。
通过生物信息学分析和qRT-PCR实验,我们的研究最终发现了6个与RA密切相关的核心基因(CD27、LCK、CD2、GZMB、IL7R和IL2RG)。这些发现可能为未来RA的临床诊断、治疗及相关研究提供有价值的方向。