Program in Computational Biology and Bioinformatics, Duke University, Durham, United States.
University of Groningen and University Medical Center Groningen, Department of Gastroenterology and Hepatology, Groningen, Netherlands.
Elife. 2023 May 9;12:e83152. doi: 10.7554/eLife.83152.
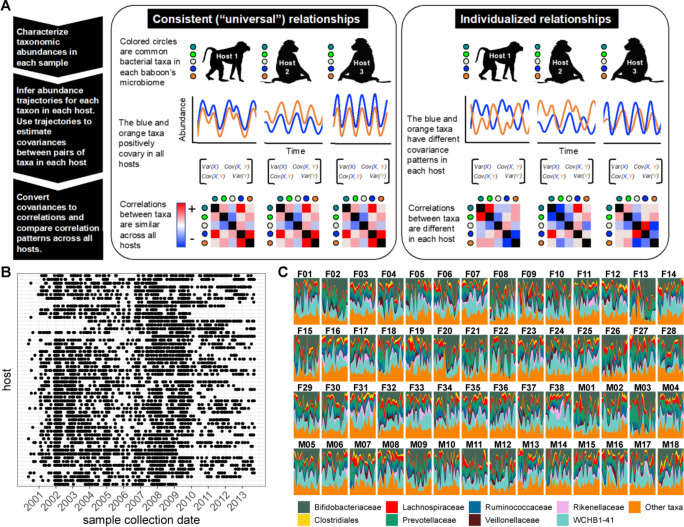
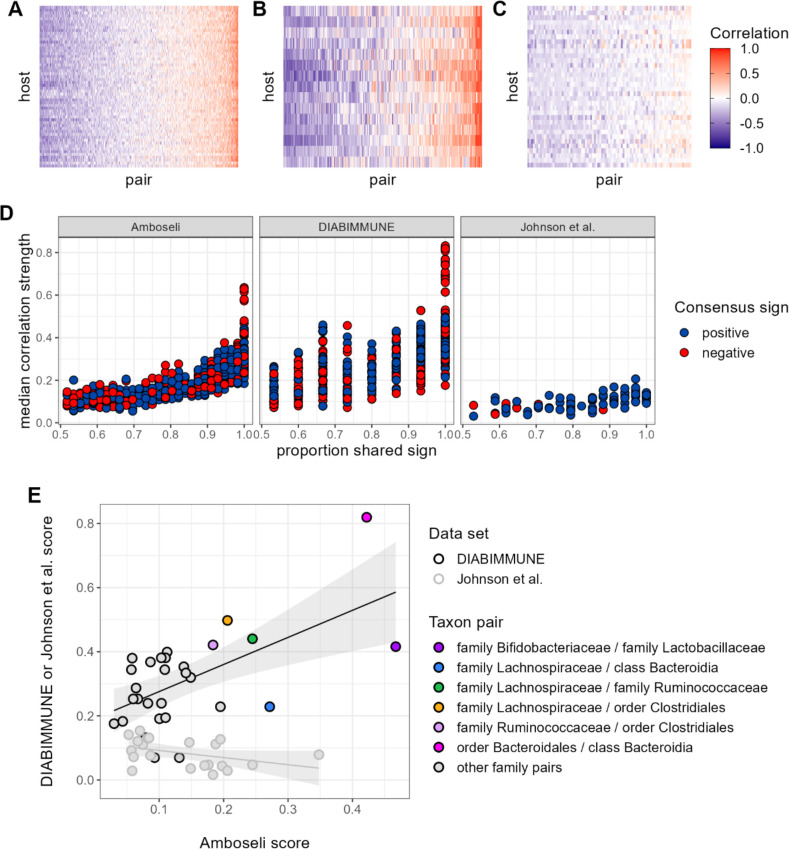
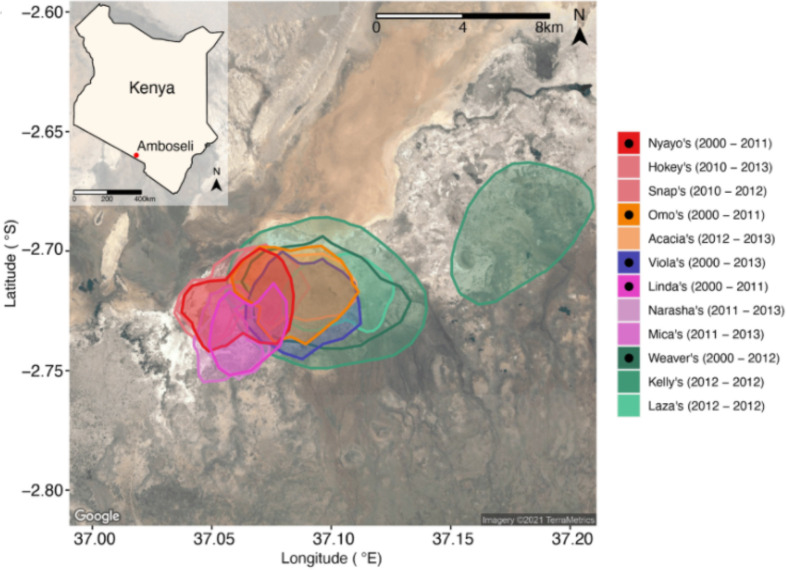
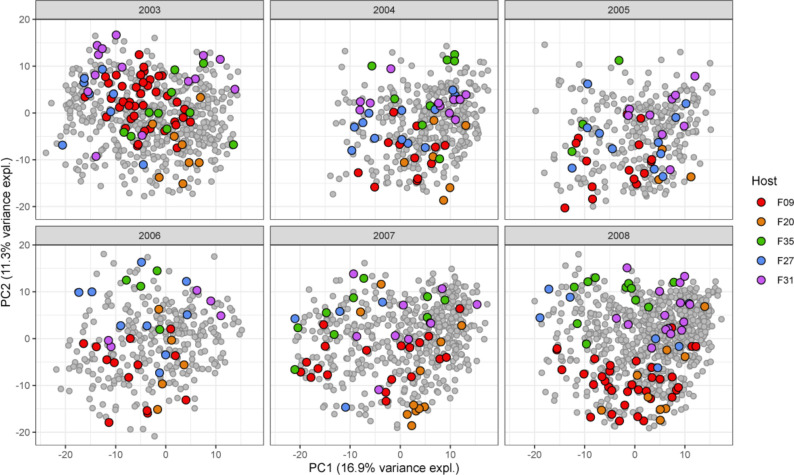
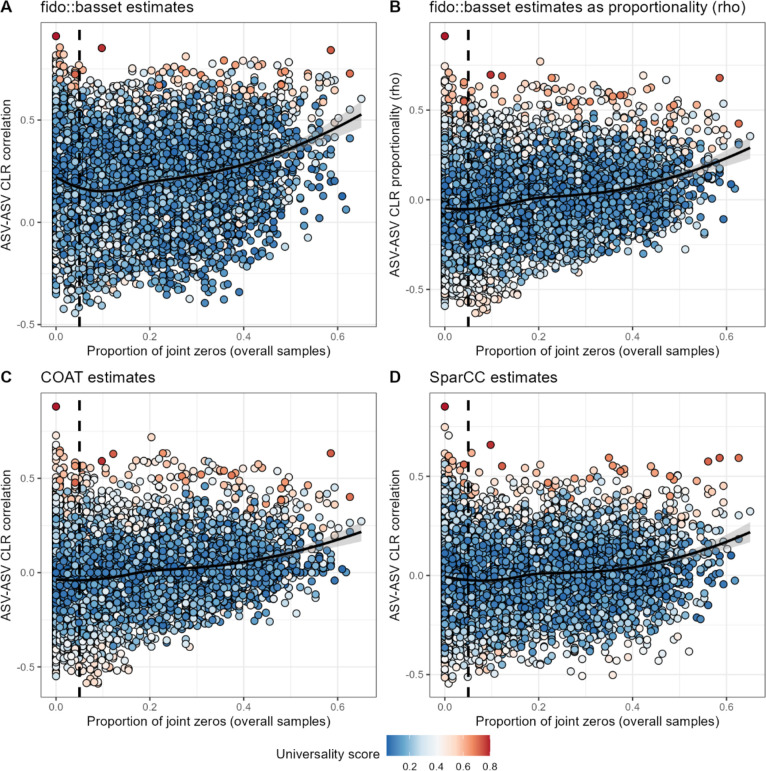
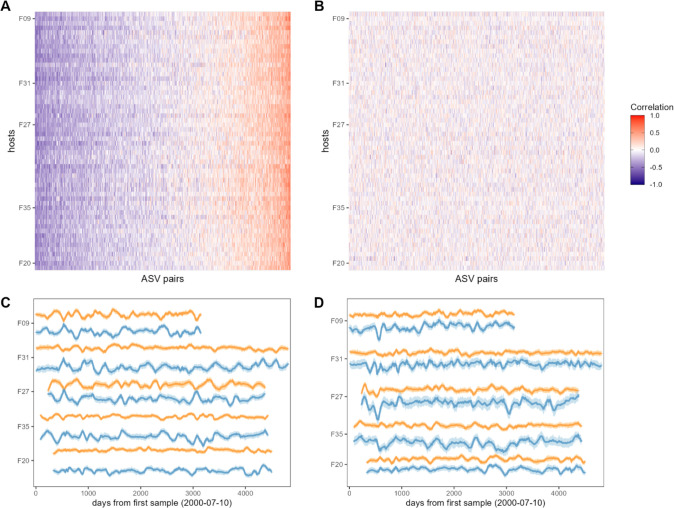
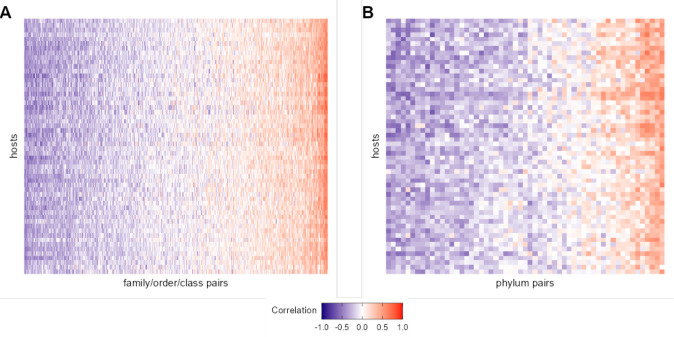
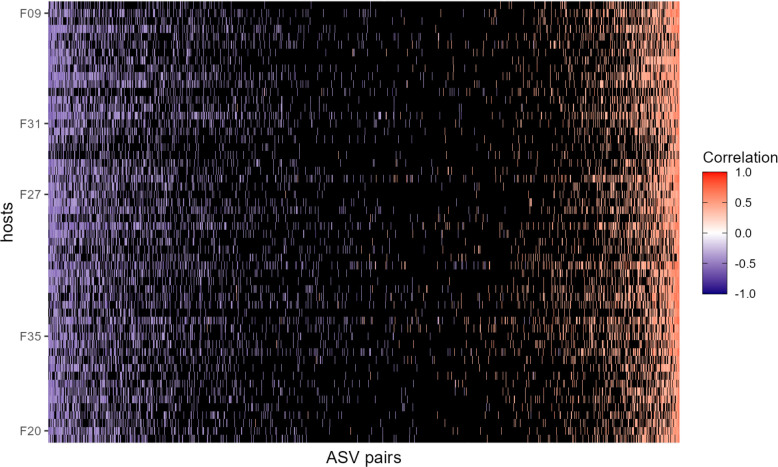
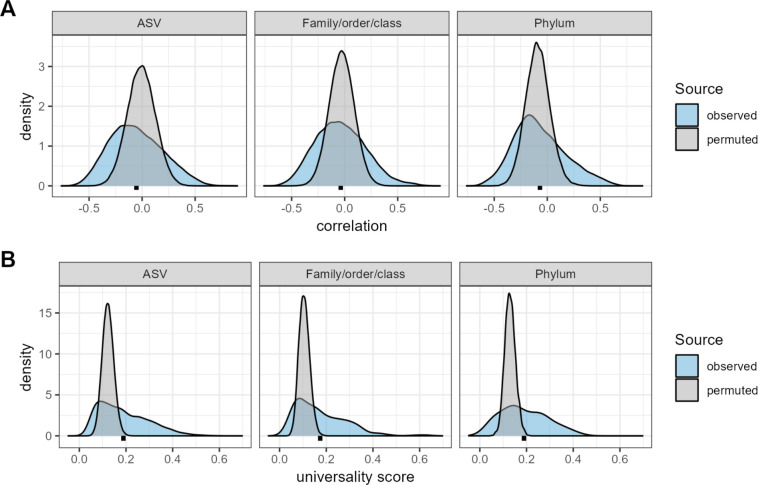
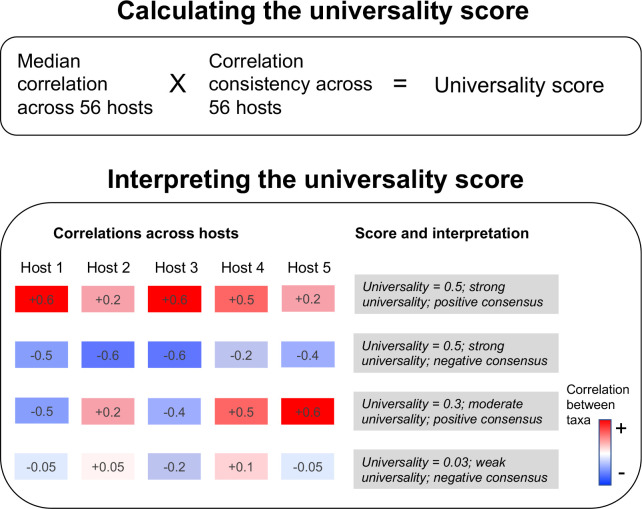
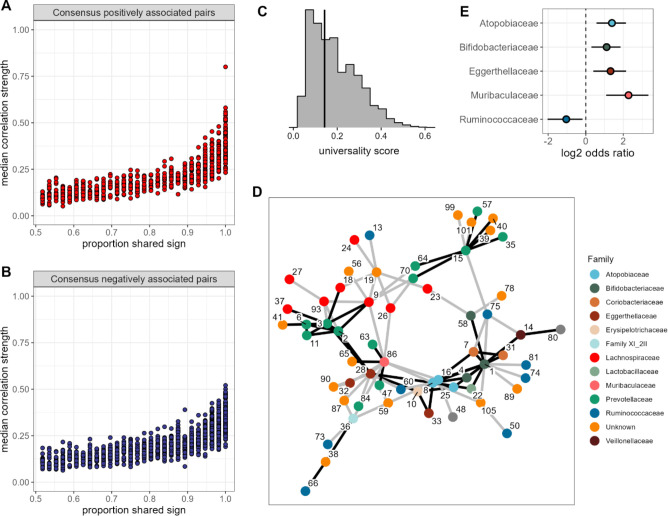
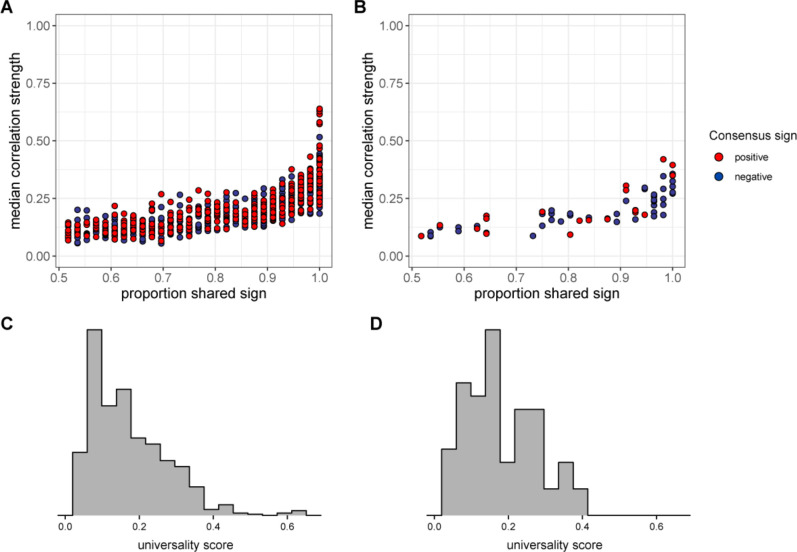
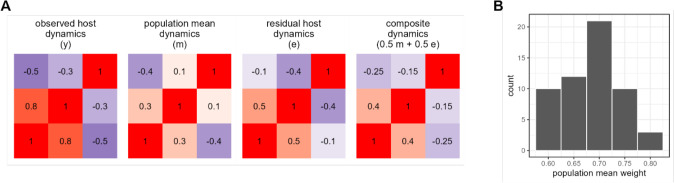
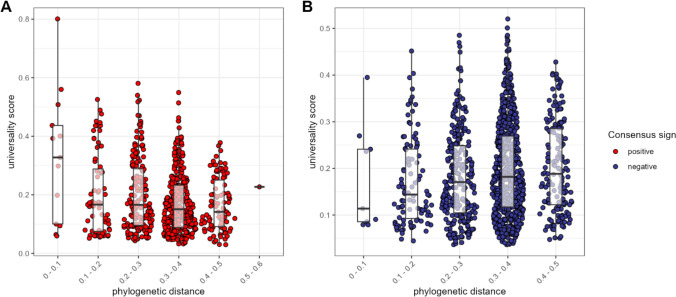
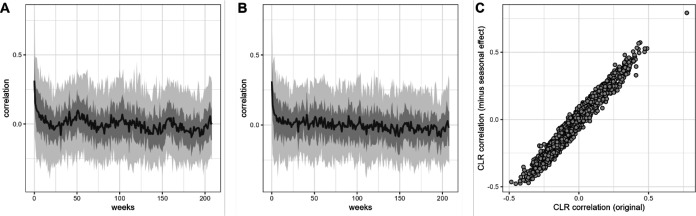
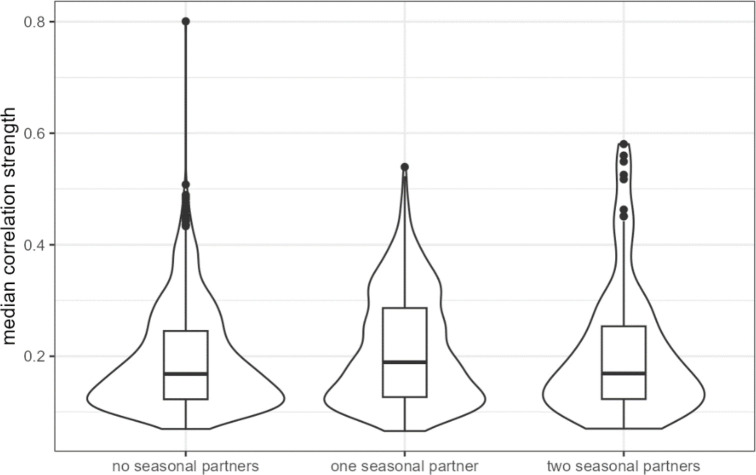
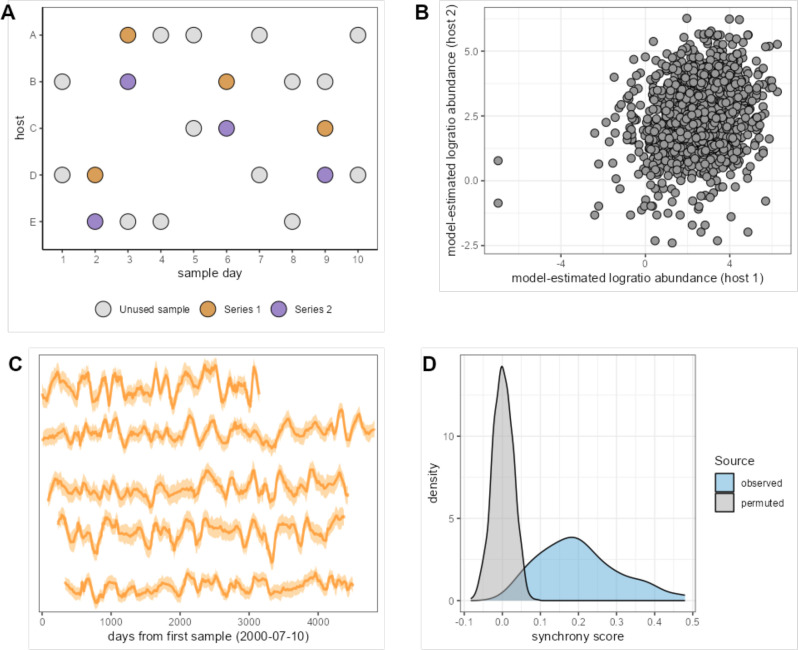
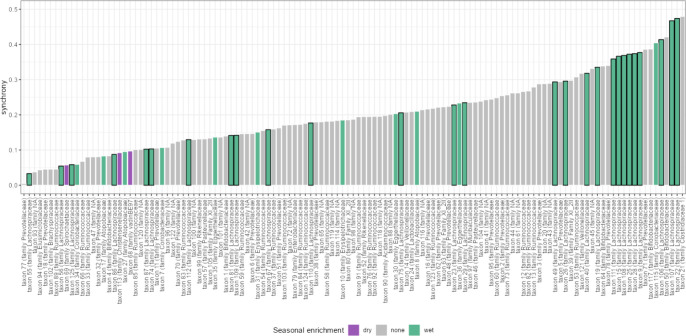
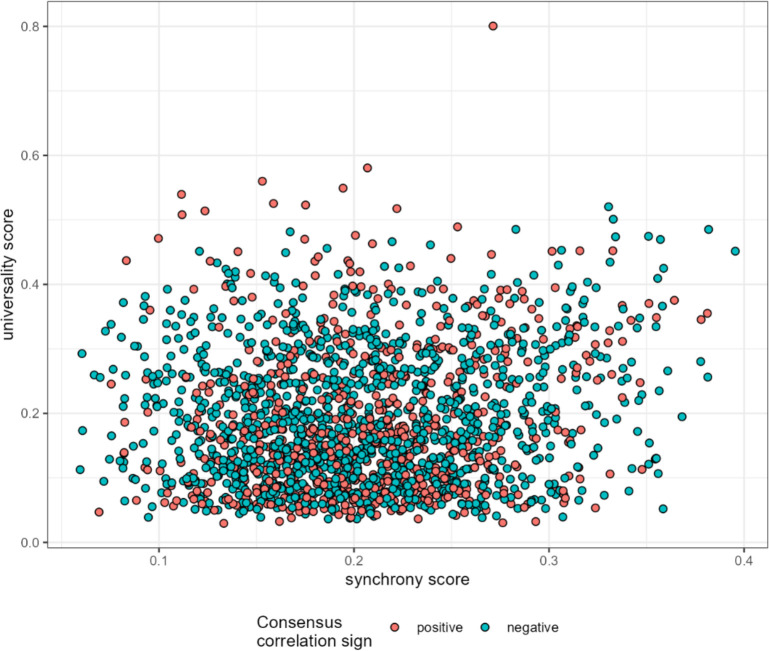
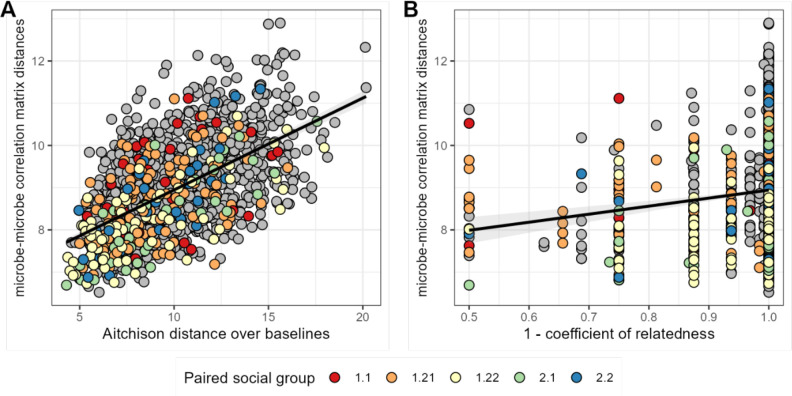
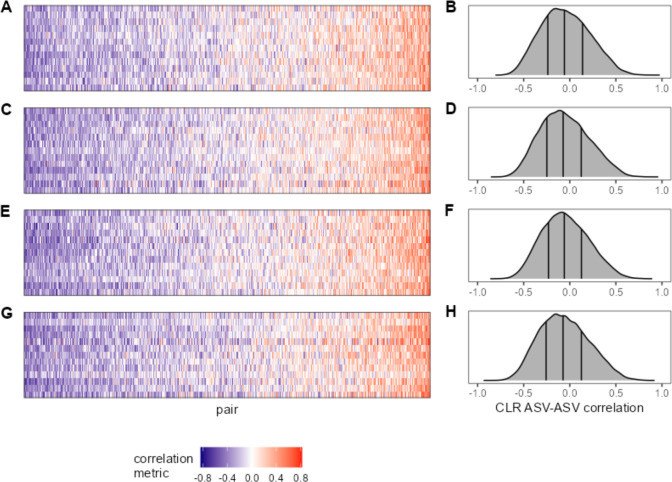
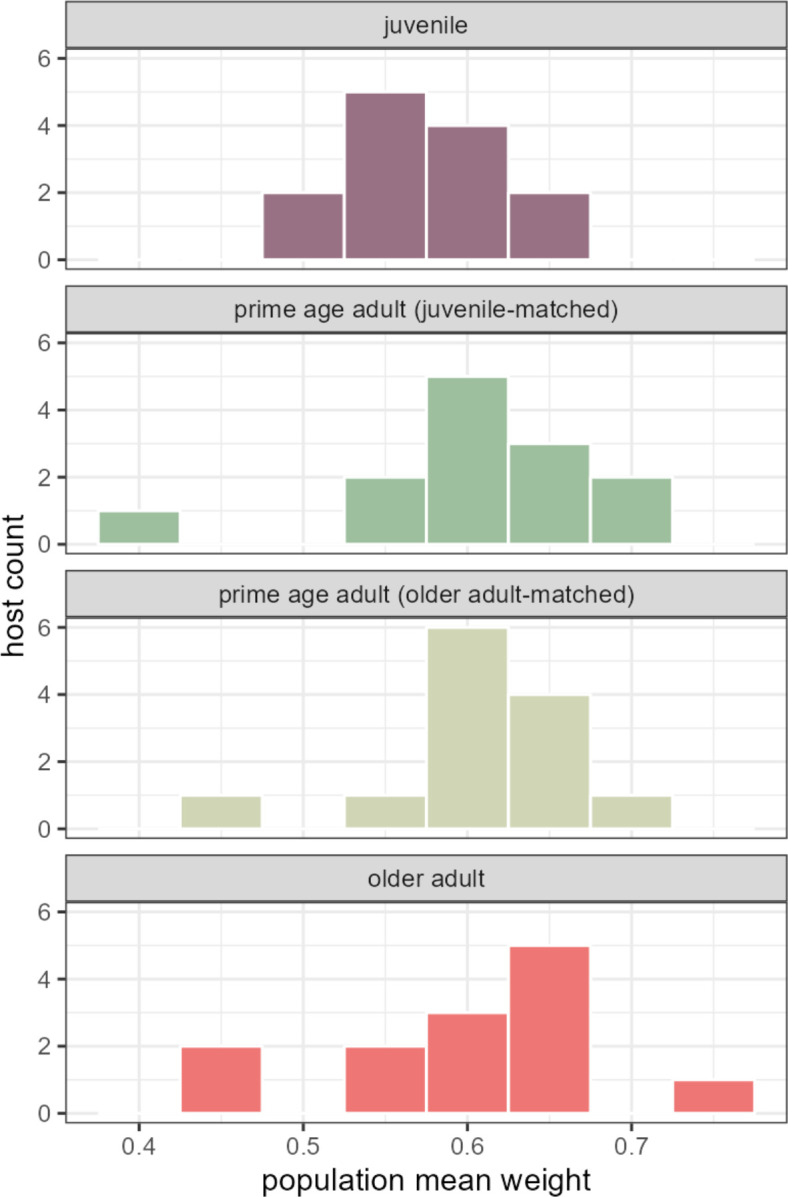
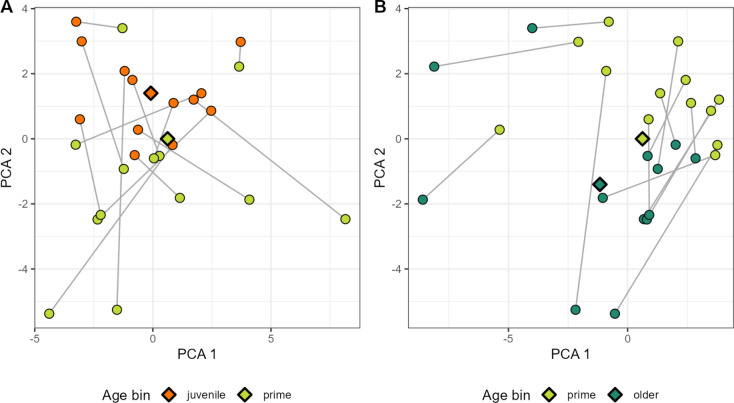
Ecological relationships between bacteria mediate the services that gut microbiomes provide to their hosts. Knowing the overall direction and strength of these relationships is essential to learn how ecology scales up to affect microbiome assembly, dynamics, and host health. However, whether bacterial relationships are generalizable across hosts or personalized to individual hosts is debated. Here, we apply a robust, multinomial logistic-normal modeling framework to extensive time series data (5534 samples from 56 baboon hosts over 13 years) to infer thousands of correlations in bacterial abundance in individual baboons and test the degree to which bacterial abundance correlations are 'universal'. We also compare these patterns to two human data sets. We find that, most bacterial correlations are weak, negative, and universal across hosts, such that shared correlation patterns dominate over host-specific correlations by almost twofold. Further, taxon pairs that had inconsistent correlation signs (either positive or negative) in different hosts always had weak correlations within hosts. From the host perspective, host pairs with the most similar bacterial correlation patterns also had similar microbiome taxonomic compositions and tended to be genetic relatives. Compared to humans, universality in baboons was similar to that in human infants, and stronger than one data set from human adults. Bacterial families that showed universal correlations in human infants were often universal in baboons. Together, our work contributes new tools for analyzing the universality of bacterial associations across hosts, with implications for microbiome personalization, community assembly, and stability, and for designing microbiome interventions to improve host health.
细菌之间的生态关系介导了肠道微生物组为其宿主提供的服务。了解这些关系的总体方向和强度对于了解生态学如何扩展以影响微生物组组装、动态和宿主健康至关重要。然而,细菌关系是否可以在宿主之间推广,或者个性化到个体宿主,这一点存在争议。在这里,我们应用了一个稳健的、多项逻辑正态建模框架,对广泛的时间序列数据(56 只狒狒宿主 13 年的 5534 个样本)进行了分析,以推断出个体狒狒中数千个细菌丰度相关性,并测试了细菌丰度相关性的普遍程度。我们还将这些模式与两个人类数据集进行了比较。我们发现,大多数细菌相关性是微弱的、负相关的,并且在宿主之间是普遍存在的,因此共享的相关模式几乎是宿主特异性相关模式的两倍。此外,在不同宿主中具有不一致相关符号(正相关或负相关)的分类群对在宿主内总是具有较弱的相关性。从宿主的角度来看,具有最相似细菌相关性模式的宿主对也具有相似的微生物组分类组成,并且往往是遗传亲属。与人类相比,狒狒的普遍性与人类婴儿相似,并且比来自人类成年人的一个数据集更强。在人类婴儿中显示出普遍相关性的细菌科通常在狒狒中也具有普遍性。总的来说,我们的工作为分析宿主间细菌关联的普遍性提供了新的工具,这对微生物组个性化、群落组装和稳定性以及设计改善宿主健康的微生物组干预措施具有重要意义。