Department of Mathematics, Stockholm University, 106 91 Stockholm, Sweden.
Department of Mathematics, Stockholm University, 106 91 Stockholm, Sweden
Genome Res. 2023 Jul;33(7):1162-1174. doi: 10.1101/gr.277645.123. Epub 2023 May 22.
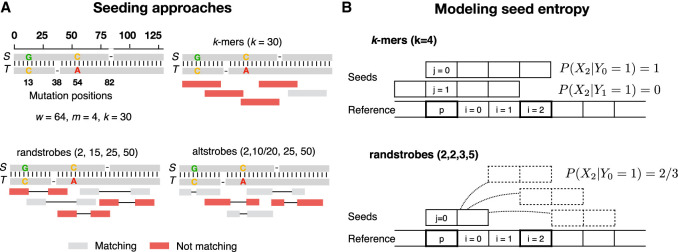
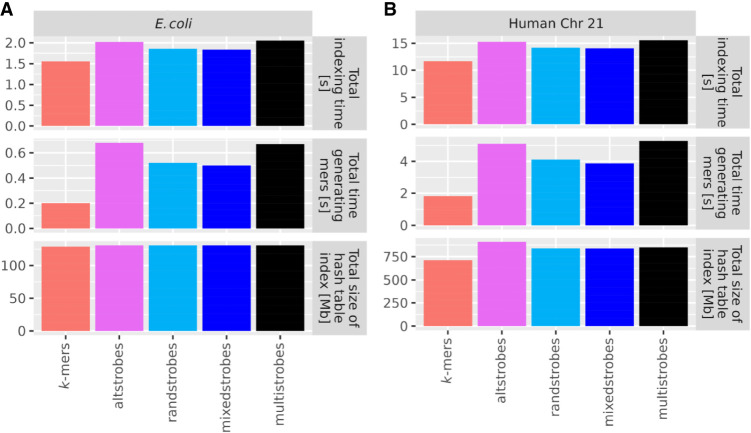
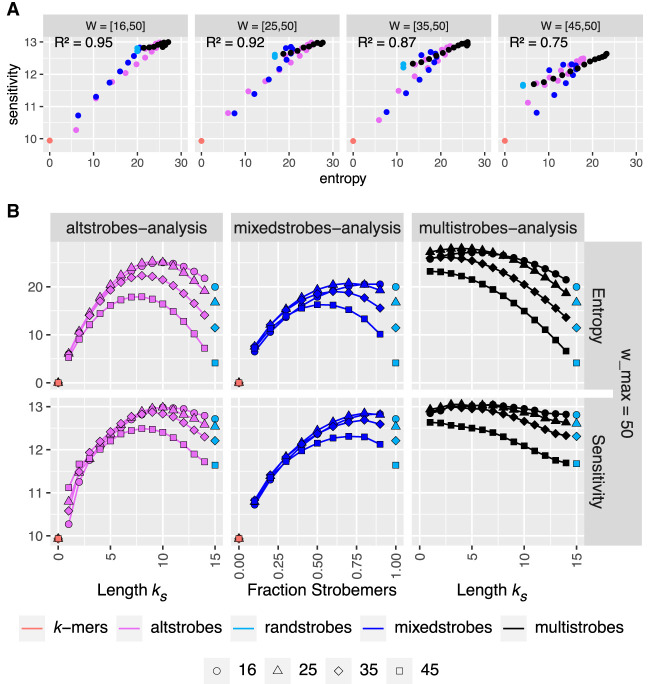
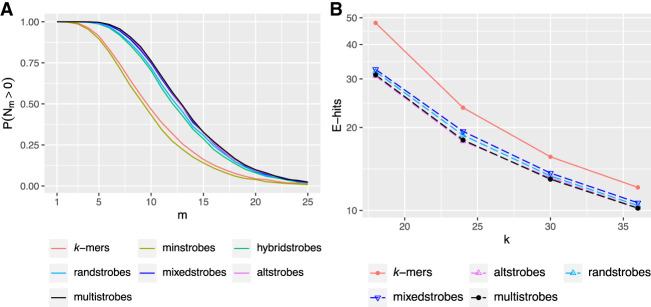
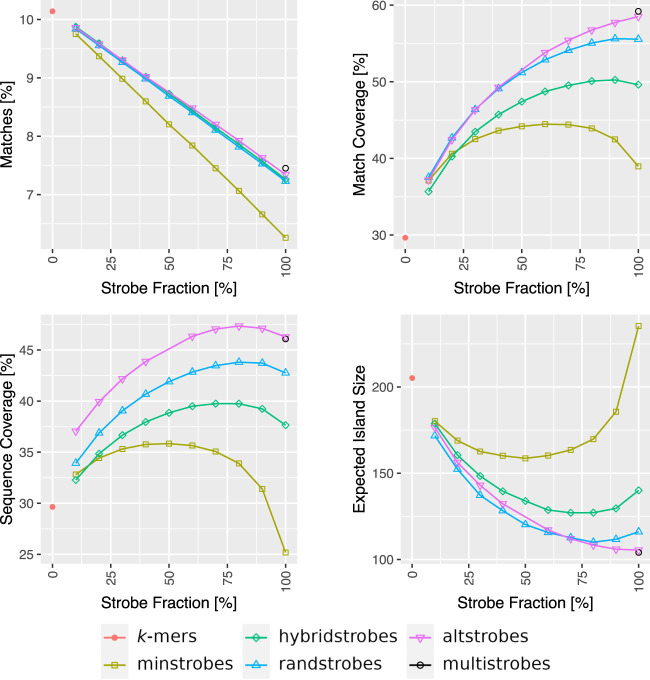
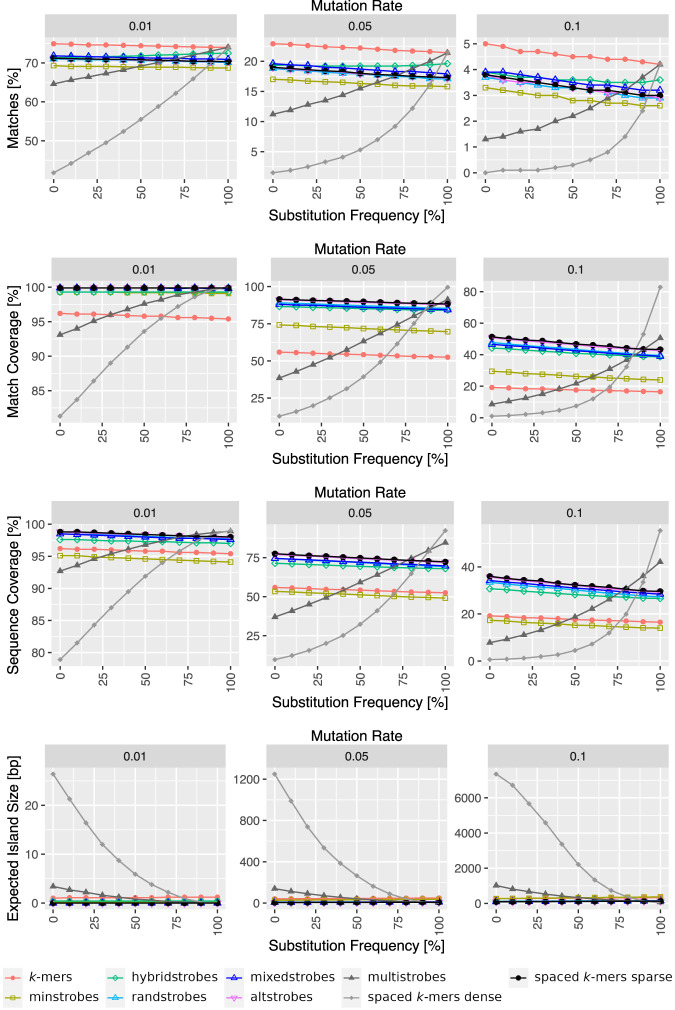
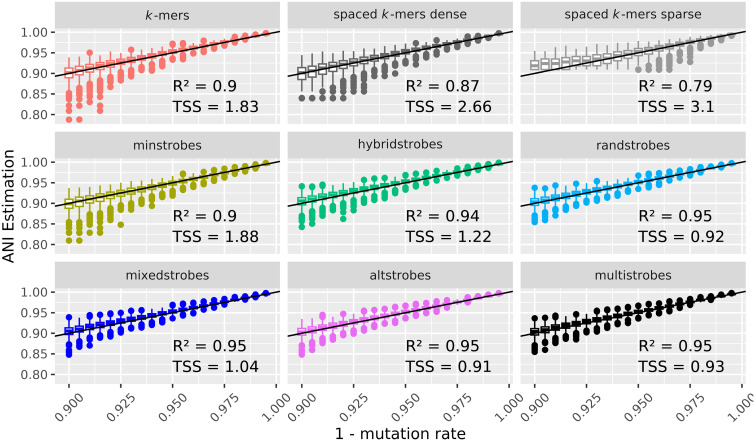
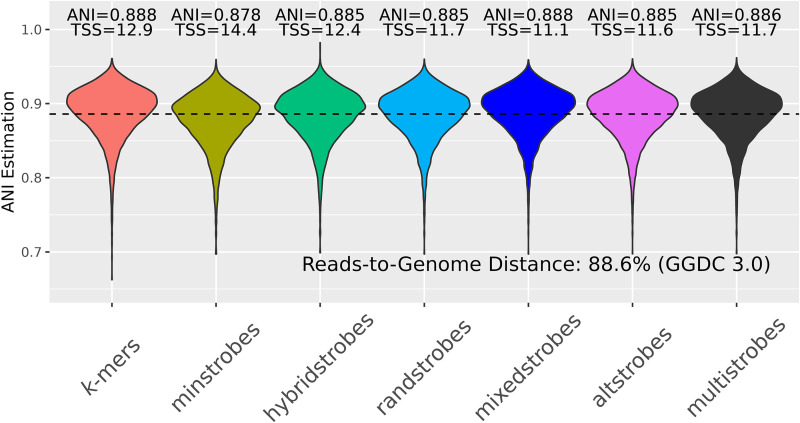
Seed design is important for sequence similarity search applications such as read mapping and average nucleotide identity (ANI) estimation. Although -mers and spaced -mers are likely the most well-known and used seeds, sensitivity suffers at high error rates, particularly when indels are present. Recently, we developed a pseudorandom seeding construct, strobemers, which was empirically shown to have high sensitivity also at high indel rates. However, the study lacked a deeper understanding of why. In this study, we propose a model to estimate the entropy of a seed and find that seeds with high entropy, according to our model, in most cases have high match sensitivity. Our discovered seed randomness-sensitivity relationship explains why some seeds perform better than others, and the relationship provides a framework for designing even more sensitive seeds. We also present three new strobemer seed constructs: mixedstrobes, altstrobes, and multistrobes. We use both simulated and biological data to show that our new seed constructs improve sequence-matching sensitivity to other strobemers. We show that the three new seed constructs are useful for read mapping and ANI estimation. For read mapping, we implement strobemers into minimap2 and observe 30% faster alignment time and 0.2% higher accuracy than using -mers when mapping reads at high error rates. As for ANI estimation, we find that higher entropy seeds have a higher rank correlation between estimated and true ANI.
种子设计对于序列相似性搜索应用程序非常重要,例如读映射和平均核苷酸同一性 (ANI) 估计。尽管 -mers 和间隔 -mers 可能是最著名和常用的种子,但在高错误率下,尤其是在存在插入和缺失的情况下,敏感性会受到影响。最近,我们开发了一种伪随机播种结构,即 strobe 种子,经验表明,即使在高插入和缺失率下,它也具有很高的敏感性。然而,该研究缺乏对原因的更深入理解。在本研究中,我们提出了一种估计种子熵的模型,并发现根据我们的模型,熵高的种子在大多数情况下具有较高的匹配敏感性。我们发现的种子随机性-敏感性关系解释了为什么某些种子比其他种子表现更好,并且该关系为设计更敏感的种子提供了框架。我们还提出了三种新的 strobe 种子结构:混合 strobe、替代 strobe 和多 strobe。我们使用模拟和生物数据表明,我们的新种子结构可以提高其他 strobe 的序列匹配敏感性。我们表明,这三种新的种子结构对于读映射和 ANI 估计都很有用。对于读映射,我们将 strobe 集成到 minimap2 中,并观察到在高错误率下映射读取时,与使用 -mers 相比,对齐时间快 30%,准确性高 0.2%。对于 ANI 估计,我们发现更高熵的种子在估计和真实 ANI 之间具有更高的等级相关系数。