Canada's Michael Smith Genome Sciences Centre, BC Cancer, Vancouver, BC, V5Z 4S6, Canada.
Bioinformatics Graduate Program, University of British Columbia, Vancouver, BC, V5Z 4S6, Canada.
Nat Commun. 2023 May 22;14(1):2940. doi: 10.1038/s41467-023-38553-y.
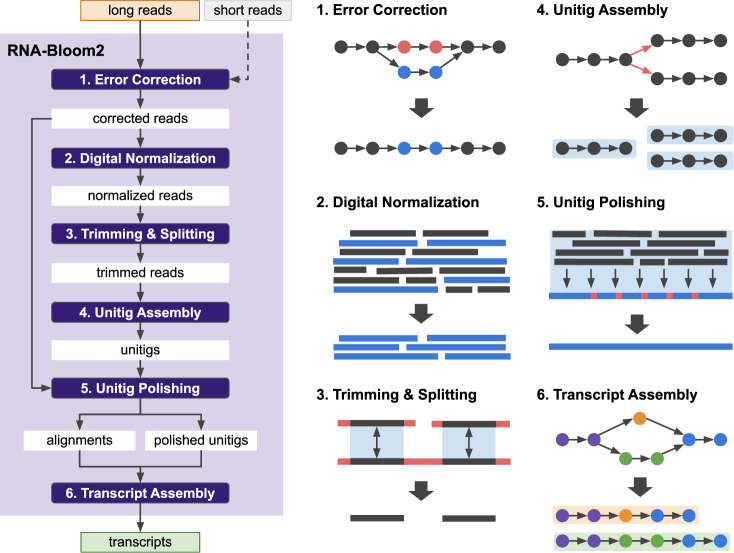
Long-read sequencing technologies have improved significantly since their emergence. Their read lengths, potentially spanning entire transcripts, is advantageous for reconstructing transcriptomes. Existing long-read transcriptome assembly methods are primarily reference-based and to date, there is little focus on reference-free transcriptome assembly. We introduce "RNA-Bloom2 [ https://github.com/bcgsc/RNA-Bloom ]", a reference-free assembly method for long-read transcriptome sequencing data. Using simulated datasets and spike-in control data, we show that the transcriptome assembly quality of RNA-Bloom2 is competitive to those of reference-based methods. Furthermore, we find that RNA-Bloom2 requires 27.0 to 80.6% of the peak memory and 3.6 to 10.8% of the total wall-clock runtime of a competing reference-free method. Finally, we showcase RNA-Bloom2 in assembling a transcriptome sample of Picea sitchensis (Sitka spruce). Since our method does not rely on a reference, it further sets the groundwork for large-scale comparative transcriptomics where high-quality draft genome assemblies are not readily available.
长读测序技术自出现以来有了显著的改进。它们的读长,可能跨越整个转录本,有利于重建转录组。现有的长读转录组组装方法主要是基于参考的,迄今为止,很少有研究关注无参考转录组组装。我们引入了“RNA-Bloom2[ https://github.com/bcgsc/RNA-Bloom ]”,这是一种用于长读转录组测序数据的无参考组装方法。使用模拟数据集和 Spike-in 对照数据,我们表明 RNA-Bloom2 的转录组组装质量可与基于参考的方法相媲美。此外,我们发现 RNA-Bloom2 需要竞争方法的峰值内存的 27.0%到 80.6%和总运行时间的 3.6%到 10.8%。最后,我们在组装 Sitka spruce(白皮松)的转录组样本中展示了 RNA-Bloom2。由于我们的方法不依赖于参考基因组,因此它为大规模比较转录组学奠定了基础,而在大规模比较转录组学中,高质量的基因组草图组装不易获得。