Division of Biostatistics, Institute for Health & Equity, and Cancer Center, Medical College of Wisconsin, Milwaukee, WI, 53226, USA.
Center for Statistical Genetics, Gertrude H. Sergievsky Center, and the Department of Neurology, Columbia University Medical Center, New York, NY, USA.
BMC Genomics. 2023 Jun 6;24(1):303. doi: 10.1186/s12864-023-09415-0.
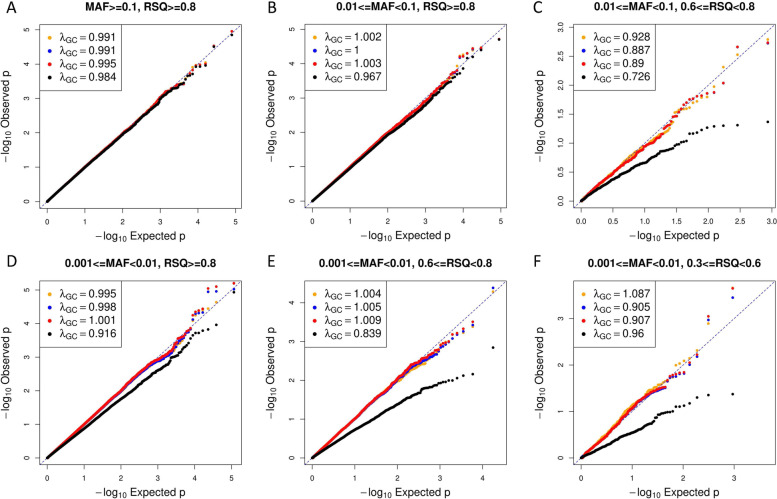
Analysis of imputed genotypes is an important and routine component of genome-wide association studies and the increasing size of imputation reference panels has facilitated the ability to impute and test low-frequency variants for associations. In the context of genotype imputation, the true genotype is unknown and genotypes are inferred with uncertainty using statistical models. Here, we present a novel method for integrating imputation uncertainty into statistical association tests using a fully conditional multiple imputation (MI) approach which is implemented using the Substantive Model Compatible Fully Conditional Specification (SMCFCS). We compared the performance of this method to an unconditional MI and two additional approaches that have been shown to demonstrate excellent performance: regression with dosages and a mixture of regression models (MRM).
Our simulations considered a range of allele frequencies and imputation qualities based on data from the UK Biobank. We found that the unconditional MI was computationally costly and overly conservative across a wide range of settings. Analyzing data with Dosage, MRM, or MI SMCFCS resulted in greater power, including for low frequency variants, compared to unconditional MI while effectively controlling type I error rates. MRM andl MI SMCFCS are both more computationally intensive then using Dosage.
The unconditional MI approach for association testing is overly conservative and we do not recommend its use in the context of imputed genotypes. Given its performance, speed, and ease of implementation, we recommend using Dosage for imputed genotypes with MAF [Formula: see text] 0.001 and Rsq [Formula: see text] 0.3.
分析推断基因型是全基因组关联研究的一个重要且常规的组成部分,越来越大的基因型推断参考面板增加了推断和测试低频变异关联的能力。在基因型推断的背景下,真实基因型是未知的,并且使用统计模型以不确定的方式推断基因型。在这里,我们提出了一种使用全条件多重推断(MI)方法将推断不确定性整合到统计关联测试中的新方法,该方法使用实质性模型兼容的全条件规范(SMCFCS)实现。我们将这种方法的性能与无条件 MI 以及另外两种已被证明具有出色性能的方法进行了比较:剂量回归和混合回归模型(MRM)。
我们的模拟考虑了基于英国生物库数据的一系列等位基因频率和推断质量。我们发现,无条件 MI 在广泛的设置中计算成本高且过于保守。与无条件 MI 相比,使用剂量、MRM 或 MI SMCFCS 分析数据可提高功效,包括对低频变异的功效,同时有效控制 I 型错误率。MRM 和 MI SMCFCS 都比使用剂量更耗费计算资源。
用于关联测试的无条件 MI 方法过于保守,我们不建议在推断基因型的情况下使用它。考虑到其性能、速度和易于实现,我们建议在 MAF [公式:见正文] 0.001 和 Rsq [公式:见正文] 0.3 的情况下使用剂量进行推断基因型。