Liu Xuwei, Zhang Yue, Li Wenjuan, Zhang Qian, Zhou Letao, Hua Yimin, Duan Hongyu, Li Yifei
Key Laboratory of Birth Defects and Related Diseases of Women and Children of MOE, Department of Pediatrics, West China Second University Hospital, Sichuan University, Chengdu, China.
Department of Nursing, West China Second University Hospital, Sichuan University, Chengdu, China.
Front Cardiovasc Med. 2023 May 23;10:1150657. doi: 10.3389/fcvm.2023.1150657. eCollection 2023.
Arrhythmogenic cardiomyopathy (ACM) is an inherited cardiomyopathy that is rarely diagnosed in infants or young children. However, some significant homozygous or compound heterozygous variants contribute to more severe clinical manifestations. In addition, inflammation of the myocardium and ventricular arrhythmia might lead to misdiagnosis with myocarditis. Here, we describe an 8-year-old patient who had been misdiagnosed with myocarditis. Timely genetic sequencing helped to identify this case as ACM induced by a homozygous variant of .
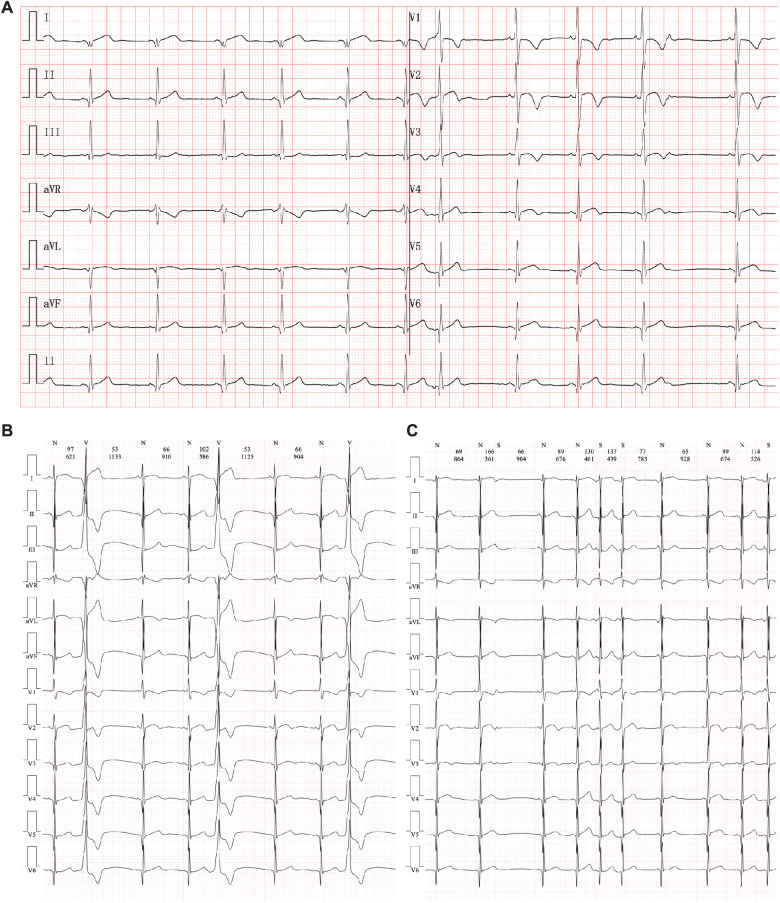
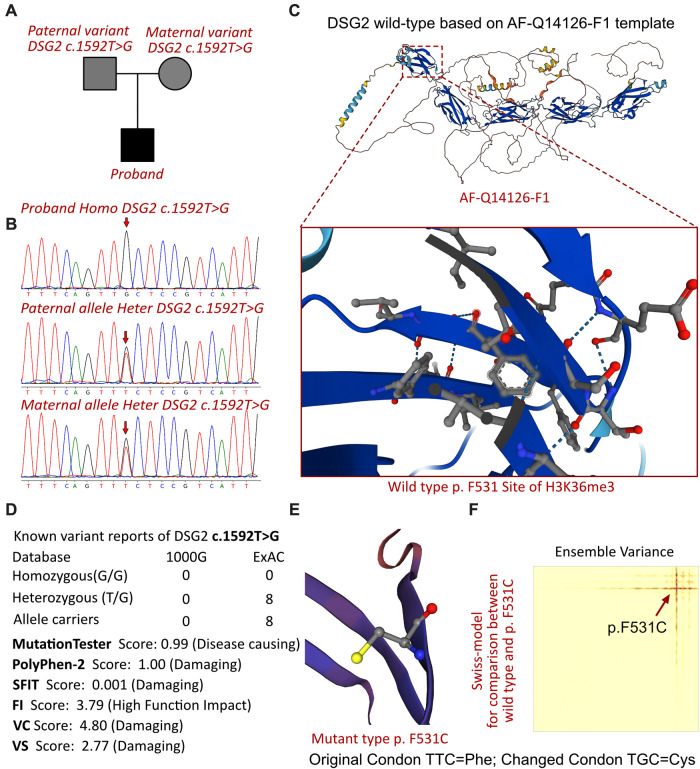
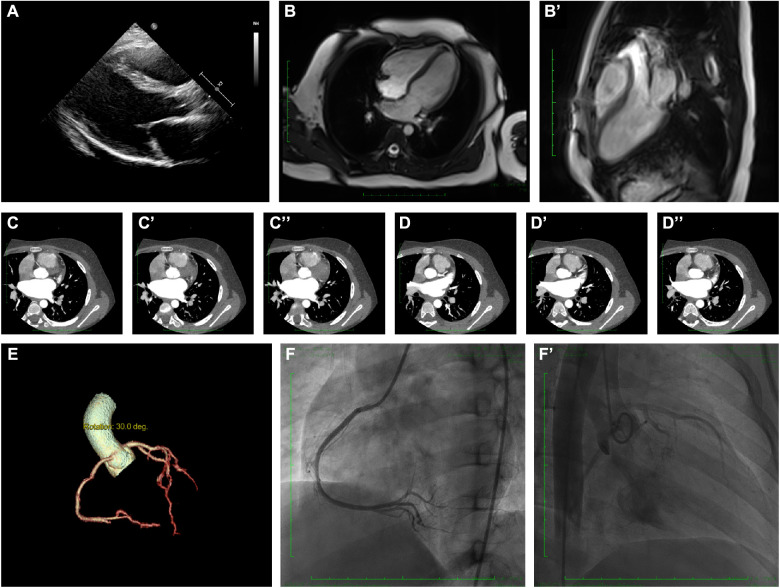
The proband of this case was an 8-year-old boy who initially presented with chest pain with an increased level of cardiac Troponin I. In addition, the electrocardiogram revealed multiple premature ventricular beats. Cardiac magnetic resonance revealed myocardial edema in the lateral ventricular wall and apex, indicating localized injuries of the myocardium. The patient was primarily suspected to have acute coronary syndrome or viral myocarditis. Whole-exome sequencing confirmed that the proband had a homozygous variation, c.1592T > G, of the gene. This mutation site was regulated by DNA modification, which induced amino acid sequence changes, protein structure effects, and splice site changes. According to MutationTaster and PolyPhen-2 analyses, the variant was considered a disease-causing mutation. Next, we used SWISS-MODEL to illustrate the mutation site of p.F531C. The ensemble variance of p.F531C indicated the free energy changes after the amino acid change.
In summary, we reported a rare pediatric case initially presenting as myocarditis that transitioned into ACM during follow-up. A homozygous genetic variant of DSG2 was inherited in the proband. This study expanded the clinical feature spectrum of DSG2-associated ACM at an early age. Additionally, the presentation of this case emphasized the difference between homozygous and heterozygous variants of desmosomal genes in disease progression. Genetic sequencing screening could be helpful in distinguishing unexplained myocarditis in children.
致心律失常性心肌病(ACM)是一种遗传性心肌病,在婴儿或幼儿中很少被诊断出来。然而,一些显著的纯合或复合杂合变异会导致更严重的临床表现。此外,心肌炎症和室性心律失常可能导致与心肌炎的误诊。在此,我们描述一名曾被误诊为心肌炎的8岁患者。及时的基因测序有助于将该病例鉴定为由 基因纯合变异引起的ACM。
该病例的先证者是一名8岁男孩,最初表现为胸痛,心肌肌钙蛋白I水平升高。此外,心电图显示多个室性早搏。心脏磁共振显示侧脑室壁和心尖部心肌水肿,提示心肌局部损伤。该患者最初被怀疑患有急性冠状动脉综合征或病毒性心肌炎。全外显子测序证实先证者的 基因存在纯合变异,c.1592T>G。该突变位点受DNA修饰调控,导致氨基酸序列改变、蛋白质结构效应和剪接位点改变。根据MutationTaster和PolyPhen-2分析,该变异被认为是致病突变。接下来,我们使用SWISS-MODEL来说明p.F531C的突变位点。p.F531C的整体变异表明氨基酸改变后的自由能变化。
总之,我们报告了一例罕见的儿科病例,最初表现为心肌炎,在随访期间转变为ACM。先证者遗传了DSG2的纯合基因变异。本研究扩展了DSG2相关ACM在幼年时的临床特征谱。此外,该病例的表现强调了桥粒基因纯合和杂合变异在疾病进展中的差异。基因测序筛查有助于区分儿童不明原因的心肌炎。