National Reference Laboratory, National Institute of Public Health, Bujumbura, Burundi.
Mohamed VI University of Health Sciences (UM6SS), Higher Institute of Biosciences and Biotechnology, Casablanca, Morocco.
BMC Genomics. 2023 Jun 10;24(1):312. doi: 10.1186/s12864-023-09420-3.
The emergence and rapid spread of new severe acute respiratory syndrome coronavirus 2 (SARS-COV-2) variants have challenged the control of the COVID-19 pandemic globally. Burundi was not spared by that pandemic, but the genetic diversity, evolution, and epidemiology of those variants in the country remained poorly understood. The present study sought to investigate the role of different SARS-COV-2 variants in the successive COVID-19 waves experienced in Burundi and the impact of their evolution on the course of that pandemic. We conducted a cross-sectional descriptive study using positive SARS-COV-2 samples for genomic sequencing. Subsequently, we performed statistical and bioinformatics analyses of the genome sequences in light of available metadata.
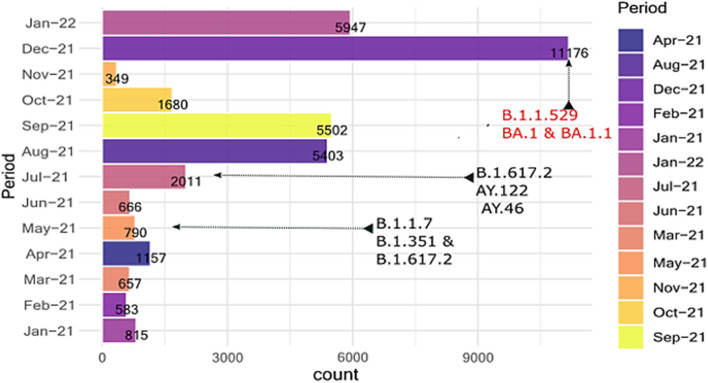
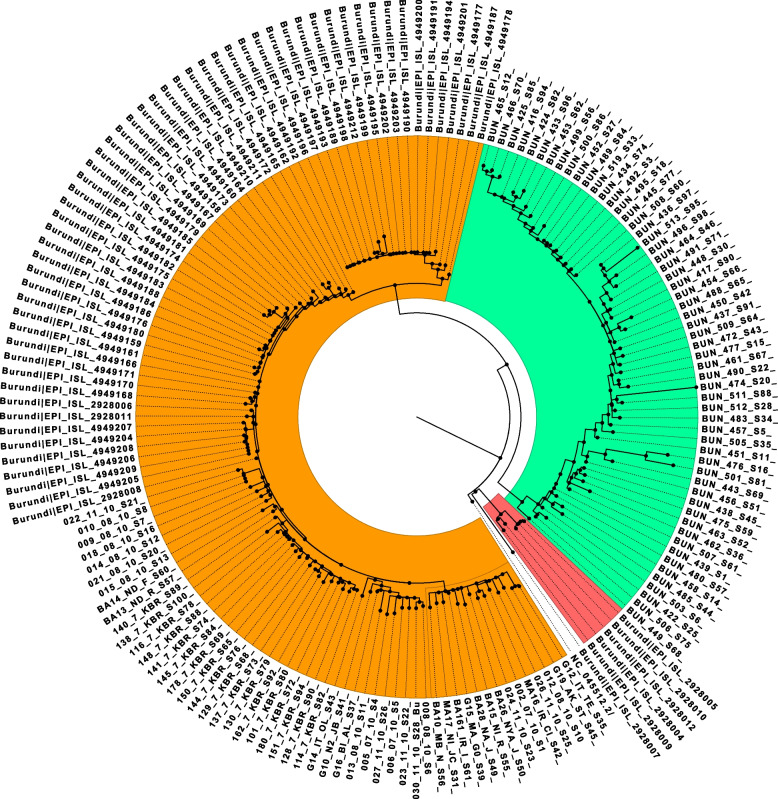
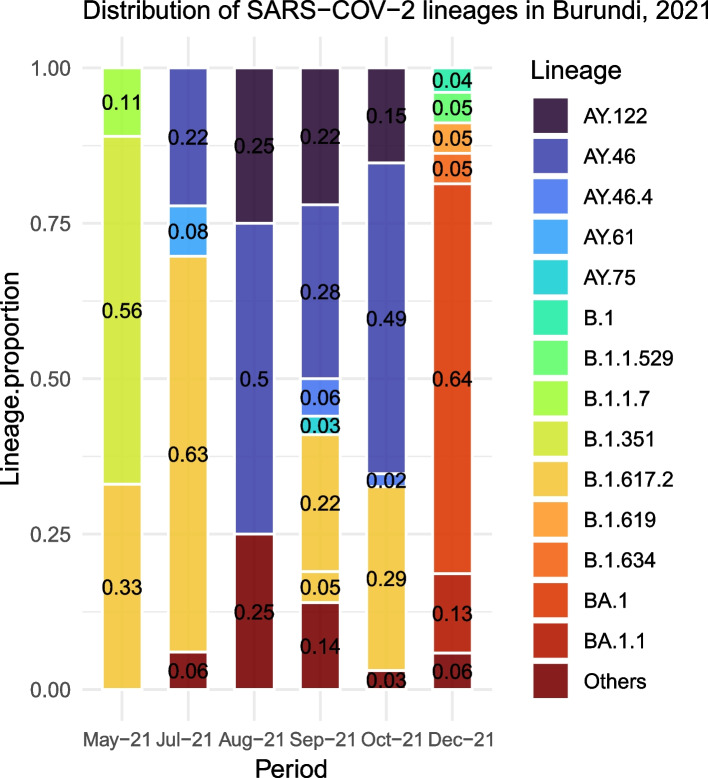
In total, we documented 27 PANGO lineages of which BA.1, B.1.617.2, AY.46, AY.122, and BA.1.1, all VOCs, accounted for 83.15% of all the genomes isolated in Burundi from May 2021 to January 2022. Delta (B.1.617.2) and its descendants predominated the peak observed in July-October 2021. It replaced the previously predominant B.1.351 lineage. It was itself subsequently replaced by Omicron (B.1.1.529, BA.1, and BA.1.1). Furthermore, we identified amino acid mutations including E484K, D614G, and L452R known to increase infectivity and immune escape in the spike proteins of Delta and Omicron variants isolated in Burundi. The SARS-COV-2 genomes from imported and community-detected cases were genetically closely related.
The global emergence of SARS-COV-2 VOCs and their subsequent introductions in Burundi was accompanied by new peaks (waves) of COVID-19. The relaxation of travel restrictions and the mutations occurring in the virus genome played an important role in the introduction and the spread of new SARS-COV-2 variants in the country. It is of utmost importance to strengthen the genomic surveillance of SARS-COV-2, enhance the protection by increasing the SARS-COV-2 vaccine coverage, and adjust the public health and social measures ahead of the emergence or introduction of new SARS-COV-2 VOCs in the country.
新型严重急性呼吸综合征冠状病毒 2 (SARS-CoV-2) 变体的出现和快速传播给全球 COVID-19 大流行的控制带来了挑战。布隆迪也未能幸免于这场大流行,但该国对这些变体的遗传多样性、进化和流行病学仍知之甚少。本研究旨在调查不同 SARS-CoV-2 变体在布隆迪经历的连续 COVID-19 浪潮中的作用,以及它们的进化对大流行进程的影响。我们使用基因组测序的 SARS-CoV-2 阳性样本进行了横断面描述性研究。随后,我们根据可用元数据对基因组序列进行了统计和生物信息学分析。
总共记录了 27 个 PANGO 谱系,其中所有 VOC 的 BA.1、B.1.617.2、AY.46、AY.122 和 BA.1.1 占 2021 年 5 月至 2022 年 1 月期间在布隆迪分离的所有基因组的 83.15%。Delta(B.1.617.2)及其后代在 2021 年 7 月至 10 月观察到的高峰中占主导地位。它取代了以前占主导地位的 B.1.351 谱系。它随后被 Omicron(B.1.1.529、BA.1 和 BA.1.1)取代。此外,我们鉴定了氨基酸突变,包括 E484K、D614G 和 L452R,这些突变已知会增加 Delta 和 Omicron 变体在布隆迪分离的刺突蛋白中的感染性和免疫逃逸。输入病例和社区检测病例的 SARS-CoV-2 基因组在遗传上密切相关。
SARS-CoV-2 VOC 的全球出现及其随后在布隆迪的引入伴随着 COVID-19 的新高峰(浪潮)。旅行限制的放松和病毒基因组中发生的突变在新 SARS-CoV-2 变体在该国的引入和传播中发挥了重要作用。加强对 SARS-CoV-2 的基因组监测,通过提高 SARS-CoV-2 疫苗覆盖率来加强保护,并在新 SARS-CoV-2 VOC 出现在该国之前调整公共卫生和社会措施至关重要。