Rodriguez Luis R, Tang Soon Yew, Barboza Willy Roque, Murthy Aditi, Tomer Yaniv, Cai Tian-Quan, Iyer Swati, Chavez Katrina, Das Ujjalkumar Subhash, Ghosh Soumita, Dimopoulos Thalia, Babu Apoorva, Connelly Caitlin, FitzGerald Garret A, Beers Michael F
Pulmonary, Allergy, and Critical Care Division Department of Medicine; Perelman School of Medicine at the University of Pennsylvania; Philadelphia, PA 19104.
PENN-CHOP Lung Biology Institute; Perelman School of Medicine at the University of Pennsylvania; Philadelphia, PA 19104.
bioRxiv. 2023 Jun 7:2023.06.07.543956. doi: 10.1101/2023.06.07.543956.
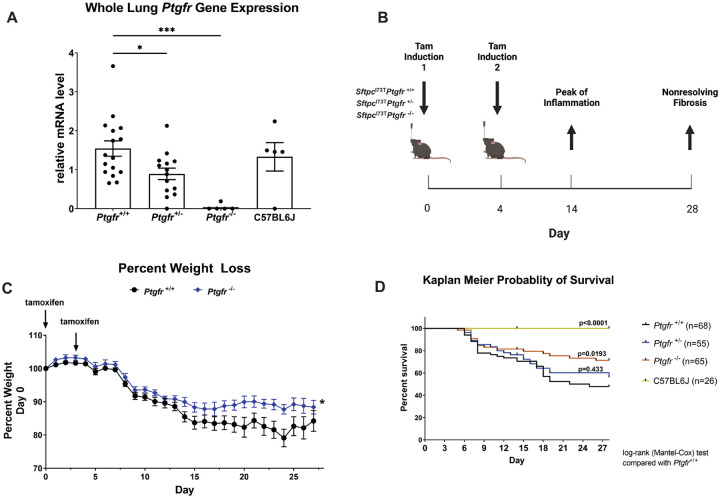
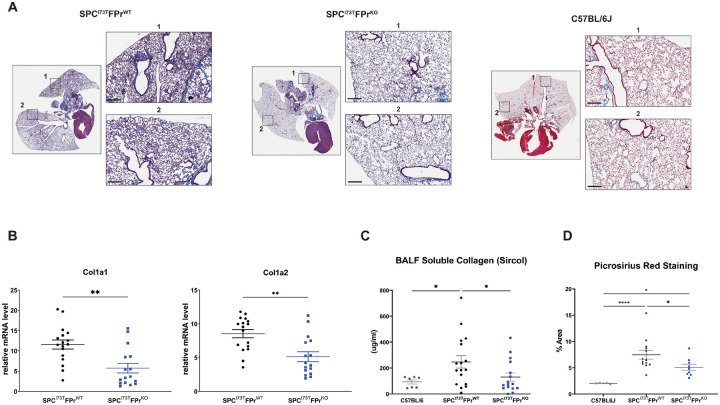
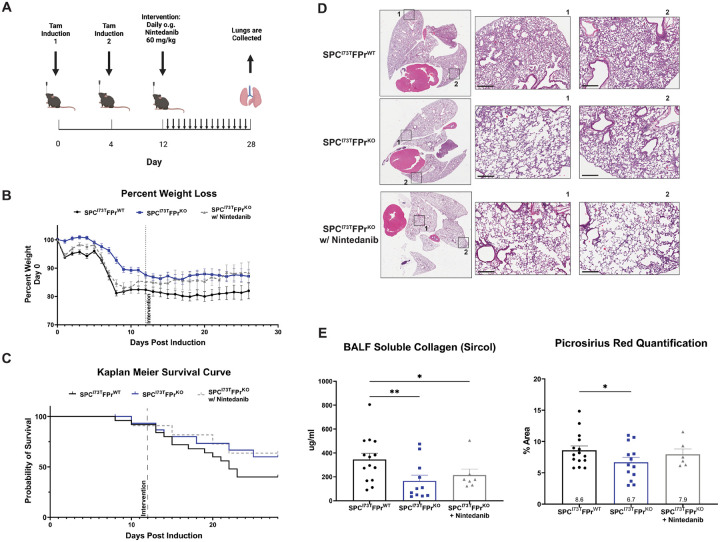
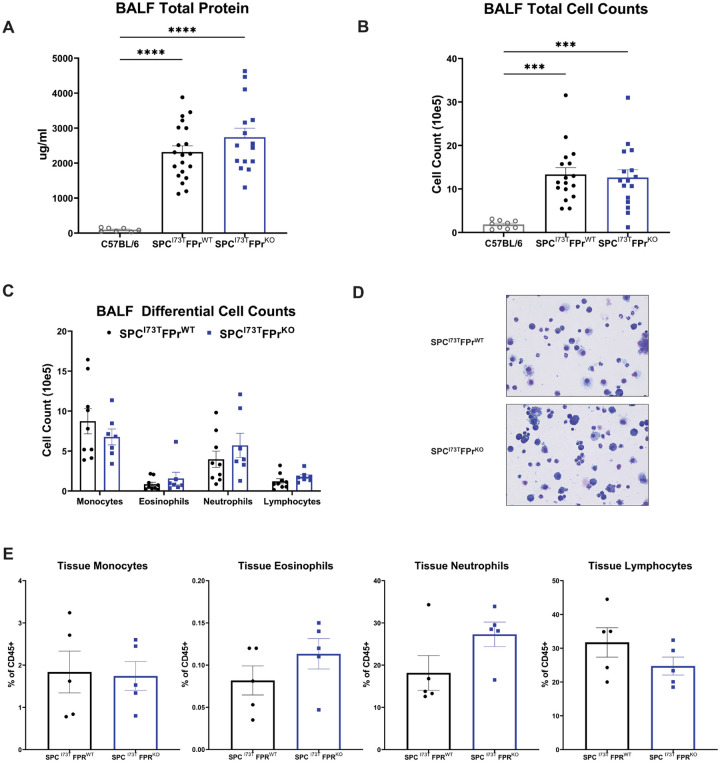
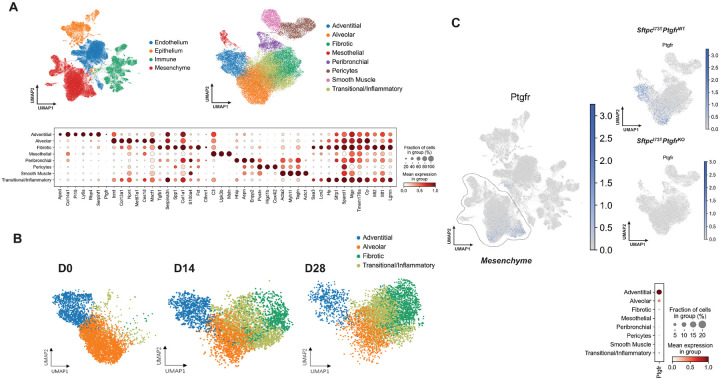
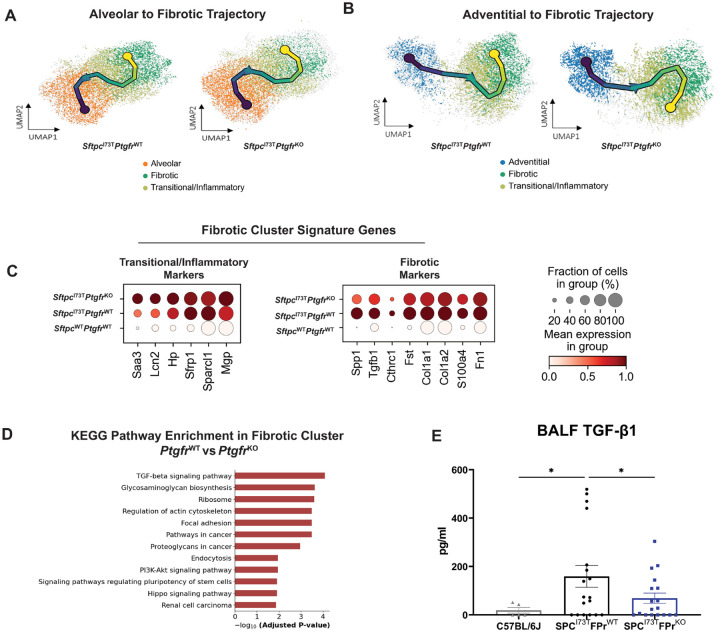
Idiopathic Pulmonary Fibrosis (IPF) is a chronic parenchymal lung disease characterized by repetitive alveolar cell injury, myofibroblast proliferation, and excessive extracellular matrix deposition for which unmet need persists for effective therapeutics. The bioactive eicosanoid, prostaglandin F2, and its cognate receptor FPr () are implicated as a TGF1 independent signaling hub for IPF. To assess this, we leveraged our published murine PF model (I - ) expressing a disease-associated missense mutation in the surfactant protein C () gene. Tamoxifen treated I- mice develop an early multiphasic alveolitis and transition to spontaneous fibrotic remodeling by 28 days. I- mice crossed to a Ptgfr null (FPr/) line showed attenuated weight loss and gene dosage dependent rescue of mortality compared to FPr+/+ cohorts. I-/FPr/ mice also showed reductions in multiple fibrotic endpoints for which administration of nintedanib was not additive. Single cell RNA sequencing, pseudotime analysis, and in vitro assays demonstrated expression predominantly within adventitial fibroblasts which were reprogrammed to an "inflammatory/transitional" cell state in a PGF2/FPr dependent manner. Collectively, the findings provide evidence for a role for PGF2 signaling in IPF, mechanistically identify a susceptible fibroblast subpopulation, and establish a benchmark effect size for disruption of this pathway in mitigating fibrotic lung remodeling.
特发性肺纤维化(IPF)是一种慢性实质性肺病,其特征为反复的肺泡细胞损伤、肌成纤维细胞增殖以及细胞外基质过度沉积,目前对于有效的治疗方法仍存在未满足的需求。生物活性类二十烷酸前列腺素F2及其同源受体FPr()被认为是IPF中一个不依赖转化生长因子β1(TGF-β1)的信号枢纽。为了对此进行评估,我们利用了已发表的表达表面活性蛋白C()基因中与疾病相关错义突变的小鼠肺纤维化模型(I-)。他莫昔芬处理的I-小鼠会发生早期多相肺泡炎,并在28天时转变为自发性纤维化重塑。与FPr+/+组相比,与Ptgfr基因敲除(FPr/)品系杂交的I-小鼠体重减轻减弱,且死亡率有基因剂量依赖性的挽救。I-/FPr/小鼠在多个纤维化终点指标上也有所降低,而尼达尼布的给药对此并无叠加作用。单细胞RNA测序、伪时间分析和体外试验表明,主要在外膜成纤维细胞中表达,这些细胞以依赖前列腺素F2/ FPr的方式重编程为“炎症/过渡”细胞状态。总体而言,这些发现为前列腺素F2信号在IPF中的作用提供了证据,从机制上确定了一个易感的成纤维细胞亚群,并为破坏该通路减轻肺纤维化重塑建立了一个基准效应大小。