Division of Molecular, Cell and Developmental Biology, School of Life Sciences, University of Dundee, Dundee DD1 5EH, UK.
Department of Epilepsy Genetics, Filadelfia Danish Epilepsy Centre, Dianalund 4293, Denmark.
Dis Model Mech. 2023 Jun 1;16(6). doi: 10.1242/dmm.049132. Epub 2023 Jun 19.
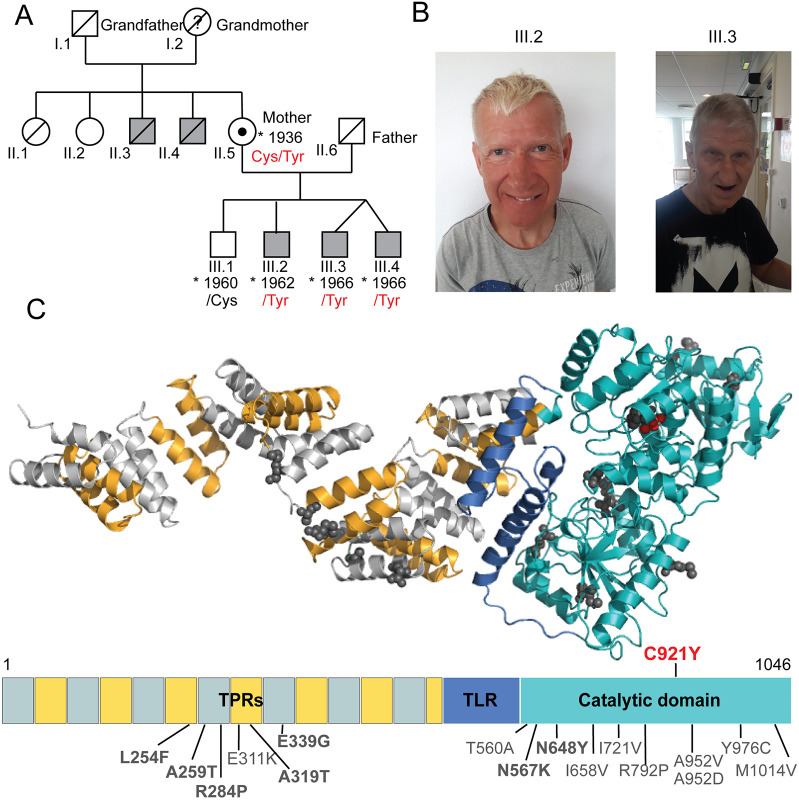
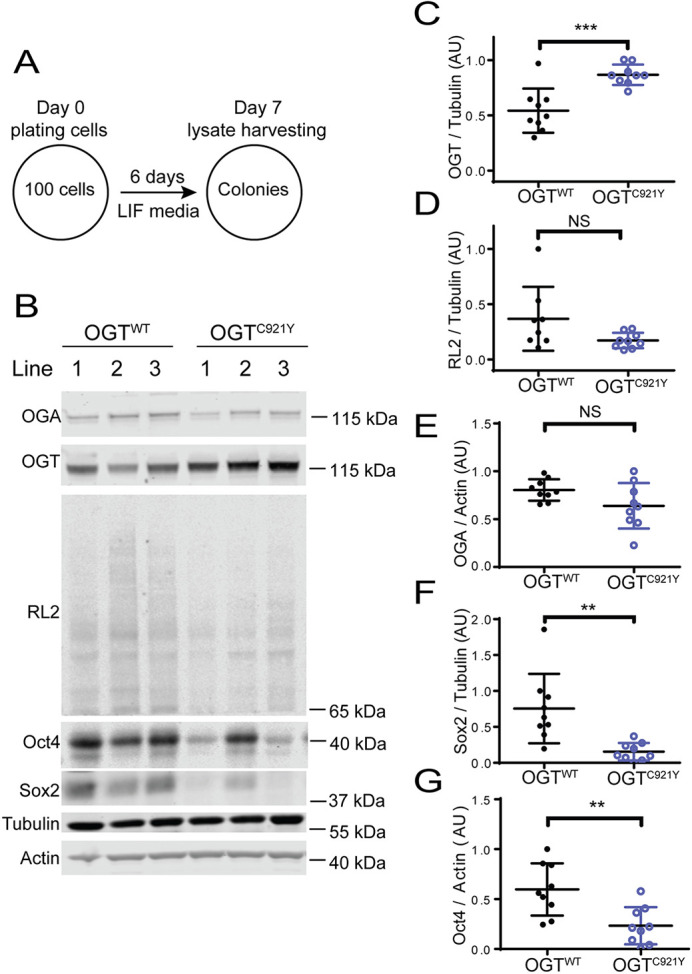
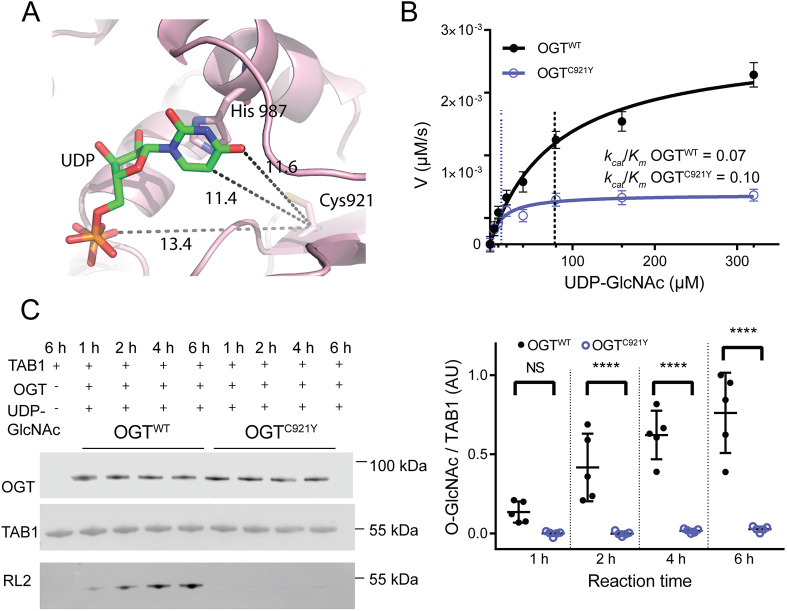
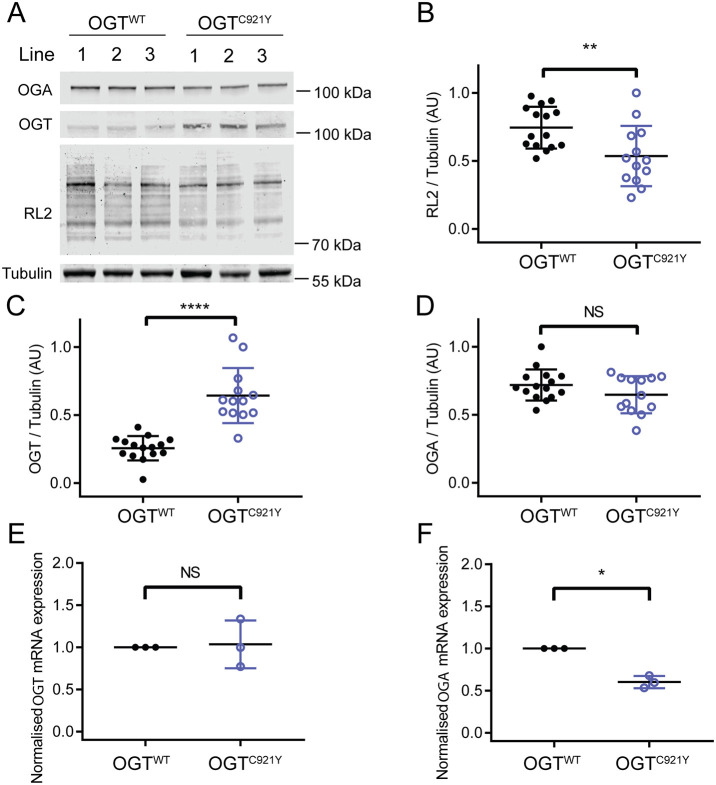
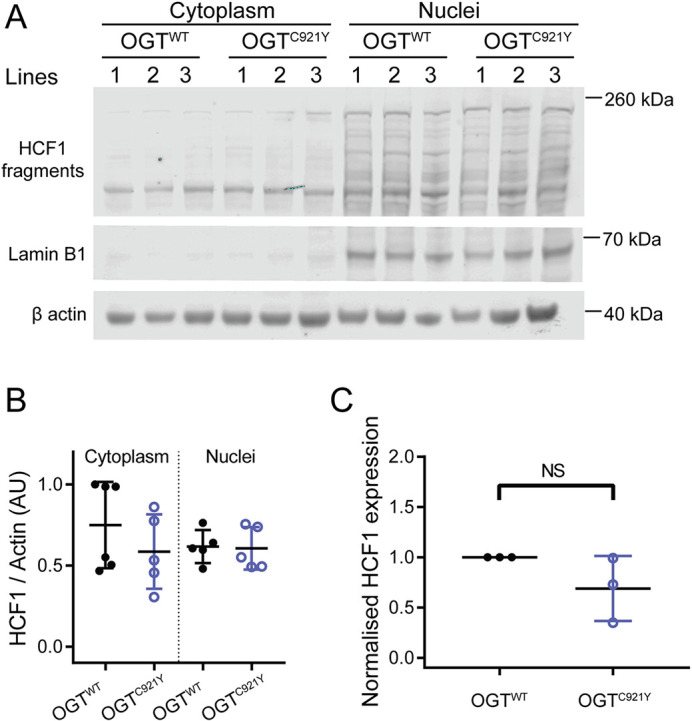
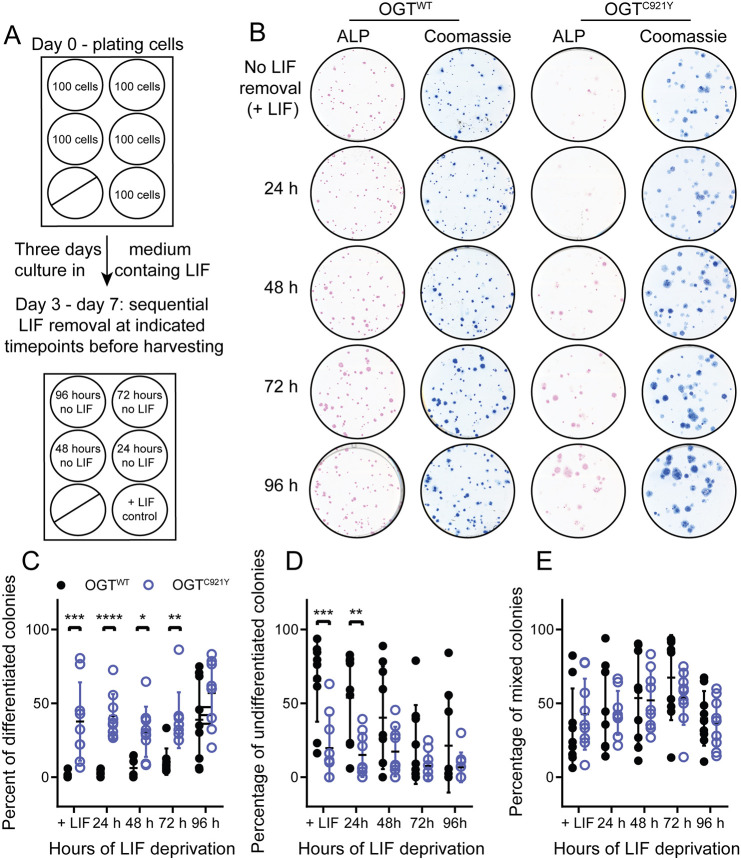
O-linked β-N-acetylglucosamine (O-GlcNAc) transferase (OGT) is an essential enzyme that modifies proteins with O-GlcNAc. Inborn OGT genetic variants were recently shown to mediate a novel type of congenital disorder of glycosylation (OGT-CDG), which is characterised by X-linked intellectual disability (XLID) and developmental delay. Here, we report an OGTC921Y variant that co-segregates with XLID and epileptic seizures, and results in loss of catalytic activity. Colonies formed by mouse embryonic stem cells carrying OGTC921Y showed decreased levels of protein O-GlcNAcylation accompanied by decreased levels of Oct4 (encoded by Pou5f1), Sox2 and extracellular alkaline phosphatase (ALP), implying reduced self-renewal capacity. These data establish a link between OGT-CDG and embryonic stem cell self-renewal, providing a foundation for examining the developmental aetiology of this syndrome.
O-连接的β-N-乙酰氨基葡萄糖(O-GlcNAc)转移酶(OGT)是一种将 O-GlcNAc 修饰到蛋白质上的必需酶。最近的研究表明,先天性 OGT 基因突变可介导一种新型的糖基化缺陷(OGT-CDG),其特征为 X 连锁智力障碍(XLID)和发育迟缓。在这里,我们报告了一个 OGTC921Y 变体,它与 XLID 和癫痫发作共分离,并导致催化活性丧失。携带 OGTC921Y 的小鼠胚胎干细胞形成的集落显示出蛋白质 O-GlcNAcylation 水平降低,同时 Oct4(由 Pou5f1 编码)、Sox2 和细胞外碱性磷酸酶(ALP)水平降低,表明自我更新能力降低。这些数据将 OGT-CDG 与胚胎干细胞自我更新联系起来,为研究该综合征的发育病因提供了基础。