Antunes Mariana A, Lopes-Pacheco Miquéias, Rocco Patricia R M
Laboratory of Pulmonary Investigation, Carlos Chagas Filho Institute of Biophysics, Federal University of Rio de Janeiro, Rio de Janeiro, RJ, Brazil.
National Institute of Science and Technology for Regenerative Medicine, Rio de Janeiro, Brazil.
Oxid Med Cell Longev. 2021 Mar 12;2021:6644002. doi: 10.1155/2021/6644002. eCollection 2021.
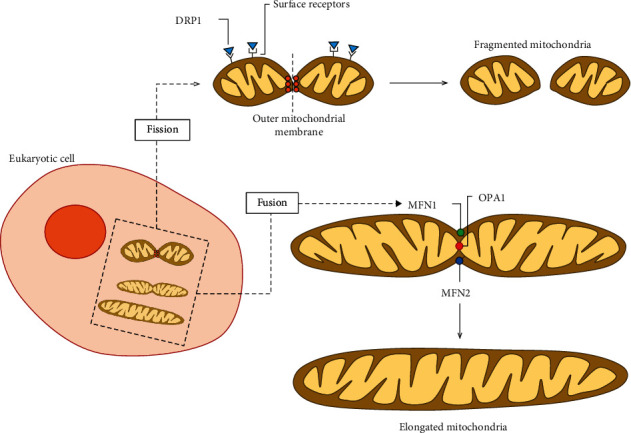
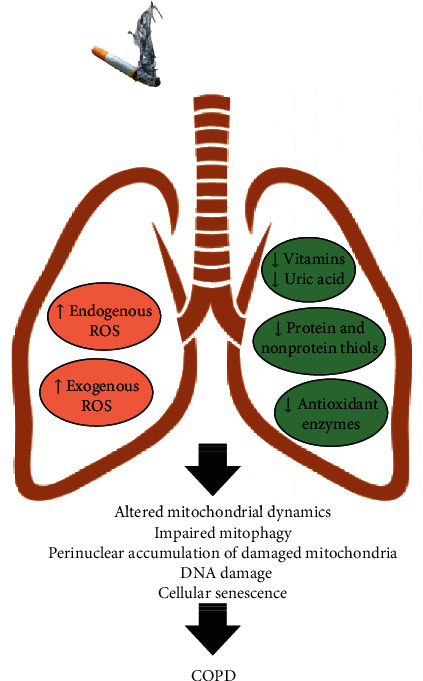
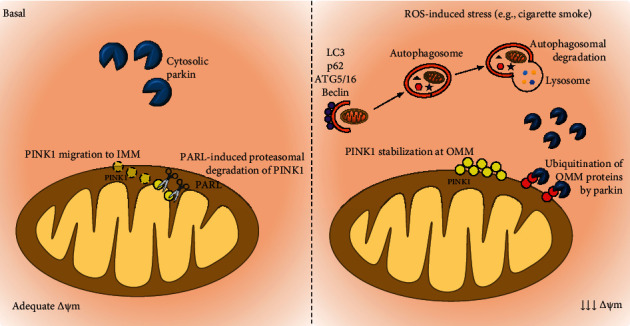
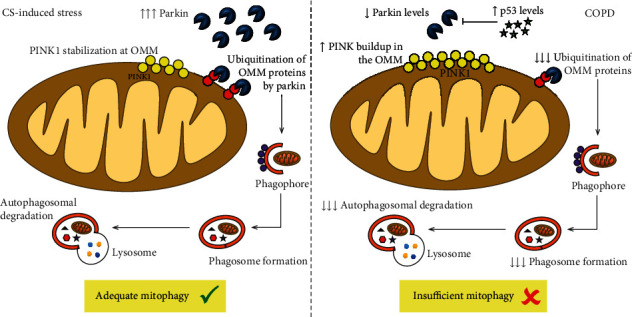
Chronic obstructive pulmonary disease (COPD) is a progressive and disabling disorder marked by airflow limitation and extensive destruction of lung parenchyma. Cigarette smoke is the major risk factor for COPD development and has been associated with increased oxidant burden on multiple cell types in the lungs. Elevated levels of reactive oxygen species (ROS) may significantly affect expression of biological molecules, signaling pathways, and function of antioxidant defenses. Although inflammatory cells, such as neutrophils and macrophages, contribute to the release of large quantities of ROS, mitochondrial dysfunction plays a critical role in ROS production due to oxidative phosphorylation. Although mitochondria are dynamic organelles, excess oxidative stress is able to alter mitochondrial function, morphology, and RNA and protein content. Indeed, mitochondria may change their shape by undergoing fusion (regulated by mitofusin 1, mitofusin 2, and optic atrophy 1 proteins) and fission (regulated by dynamin-related protein 1), which are essential processes to maintain a healthy and functional mitochondrial network. Cigarette smoke can induce mitochondrial hyperfusion, thus reducing mitochondrial quality control and cellular stress resistance. Furthermore, diminished levels of enzymes involved in the mitophagy process, such as Parkin (a ubiquitin ligase E3) and the PTEN-induced putative kinase 1 (PINK1), are commonly observed in COPD and correlate directly with faulty removal of dysfunctional mitochondria and consequent cell senescence in this disorder. In this review, we highlight the main mechanisms for the regulation of mitochondrial quality and how they are affected by oxidative stress during COPD development and progression.
慢性阻塞性肺疾病(COPD)是一种进行性致残性疾病,其特征为气流受限和肺实质的广泛破坏。香烟烟雾是COPD发生的主要危险因素,并与肺部多种细胞类型上氧化剂负担增加有关。活性氧(ROS)水平升高可能会显著影响生物分子的表达、信号通路以及抗氧化防御功能。尽管中性粒细胞和巨噬细胞等炎症细胞会促使大量ROS释放,但线粒体功能障碍在因氧化磷酸化导致的ROS产生中起关键作用。尽管线粒体是动态细胞器,但过量的氧化应激能够改变线粒体功能、形态以及RNA和蛋白质含量。事实上,线粒体可通过融合(由线粒体融合蛋白1、线粒体融合蛋白2和视神经萎缩蛋白1调控)和裂变(由动力相关蛋白1调控)来改变其形状,而这是维持健康且功能正常的线粒体网络的重要过程。香烟烟雾可诱导线粒体过度融合,从而降低线粒体质量控制和细胞应激抗性。此外,在COPD中通常会观察到参与线粒体自噬过程的酶水平降低,如帕金蛋白(一种泛素连接酶E3)和PTEN诱导激酶1(PINK1),且这与该疾病中功能失调线粒体的清除故障及随之而来的细胞衰老直接相关。在本综述中,我们着重介绍线粒体质量调控的主要机制以及它们在COPD发生发展过程中如何受到氧化应激的影响。