Computational and Systems Biology Program, Sloan Kettering Institute, Memorial Sloan Kettering Cancer Center, New York, New York 10065, United States.
Tri-Institutional PhD Program in Computational Biology and Medicine, Weill Cornell Medical College, Cornell University, New York, New York 10065, United States.
J Chem Theory Comput. 2023 Aug 8;19(15):4863-4882. doi: 10.1021/acs.jctc.3c00333. Epub 2023 Jul 14.
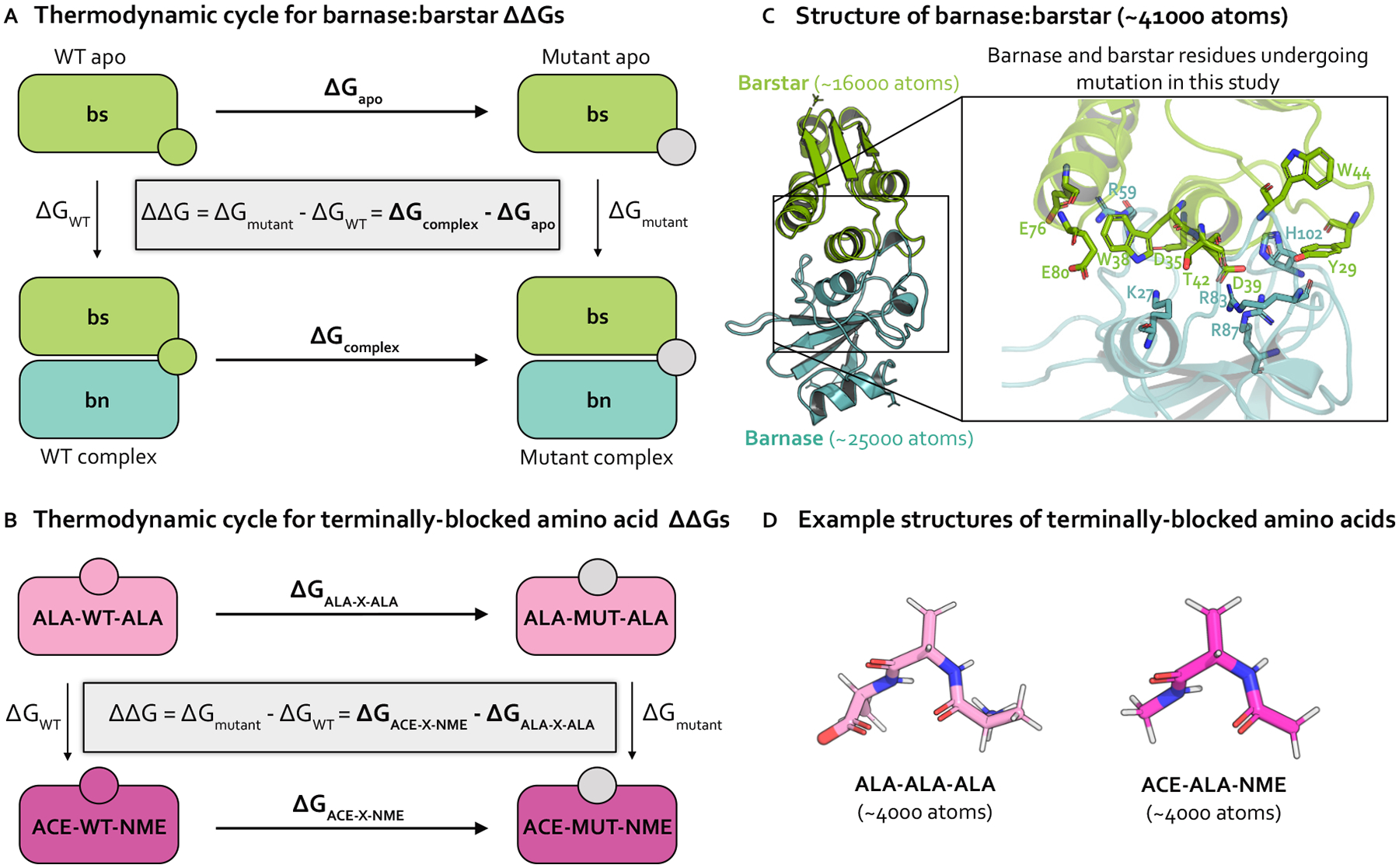
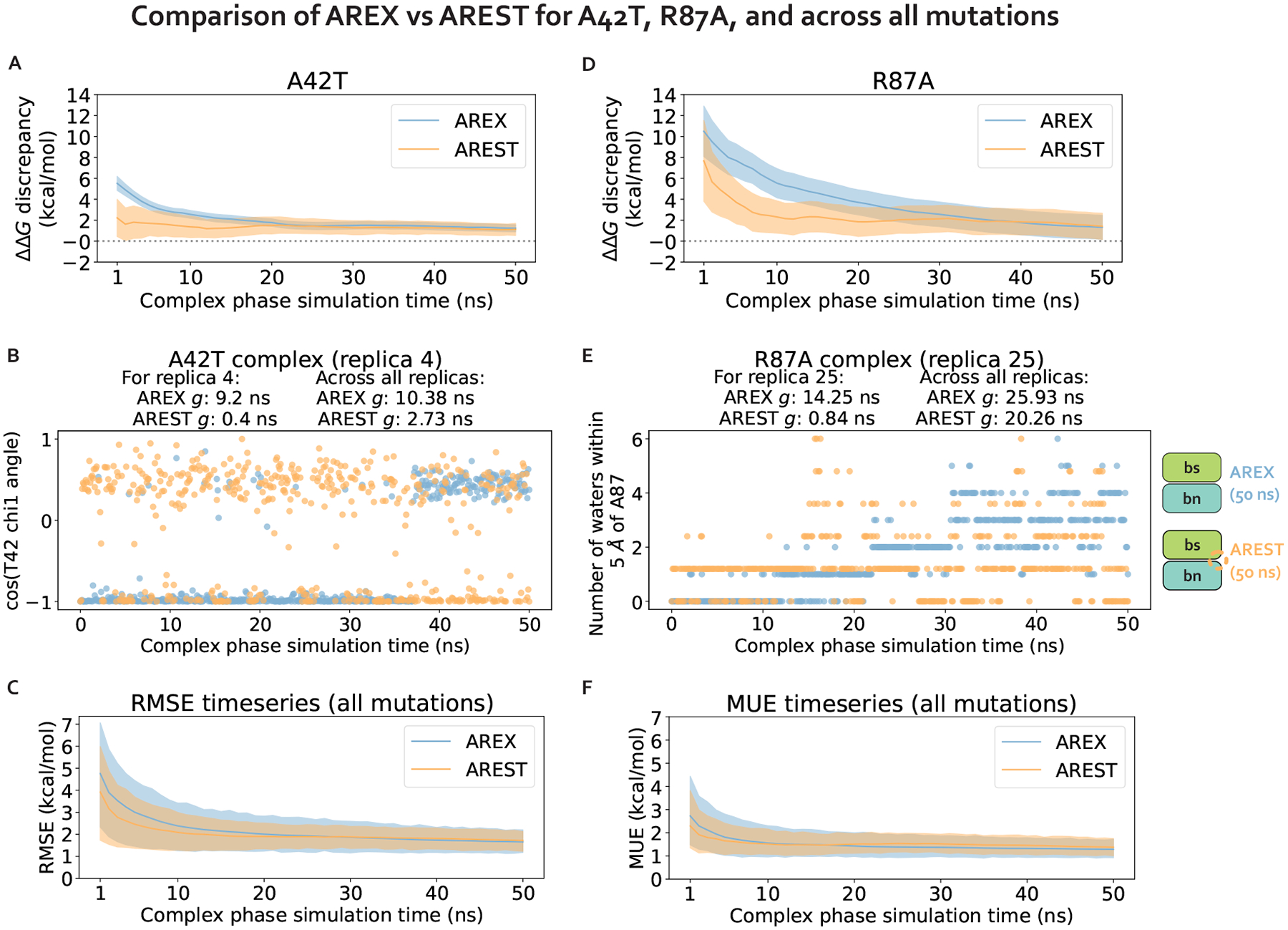
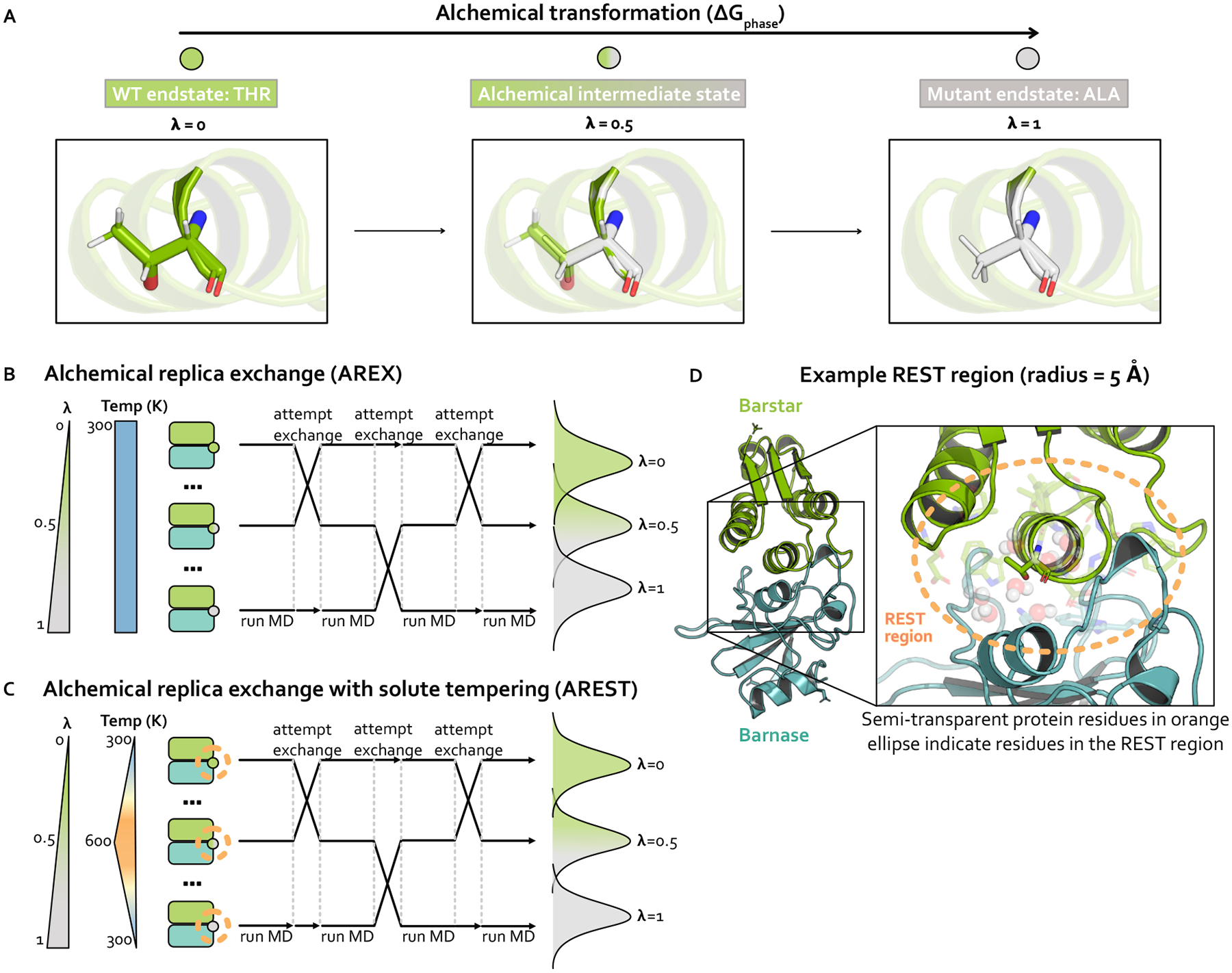
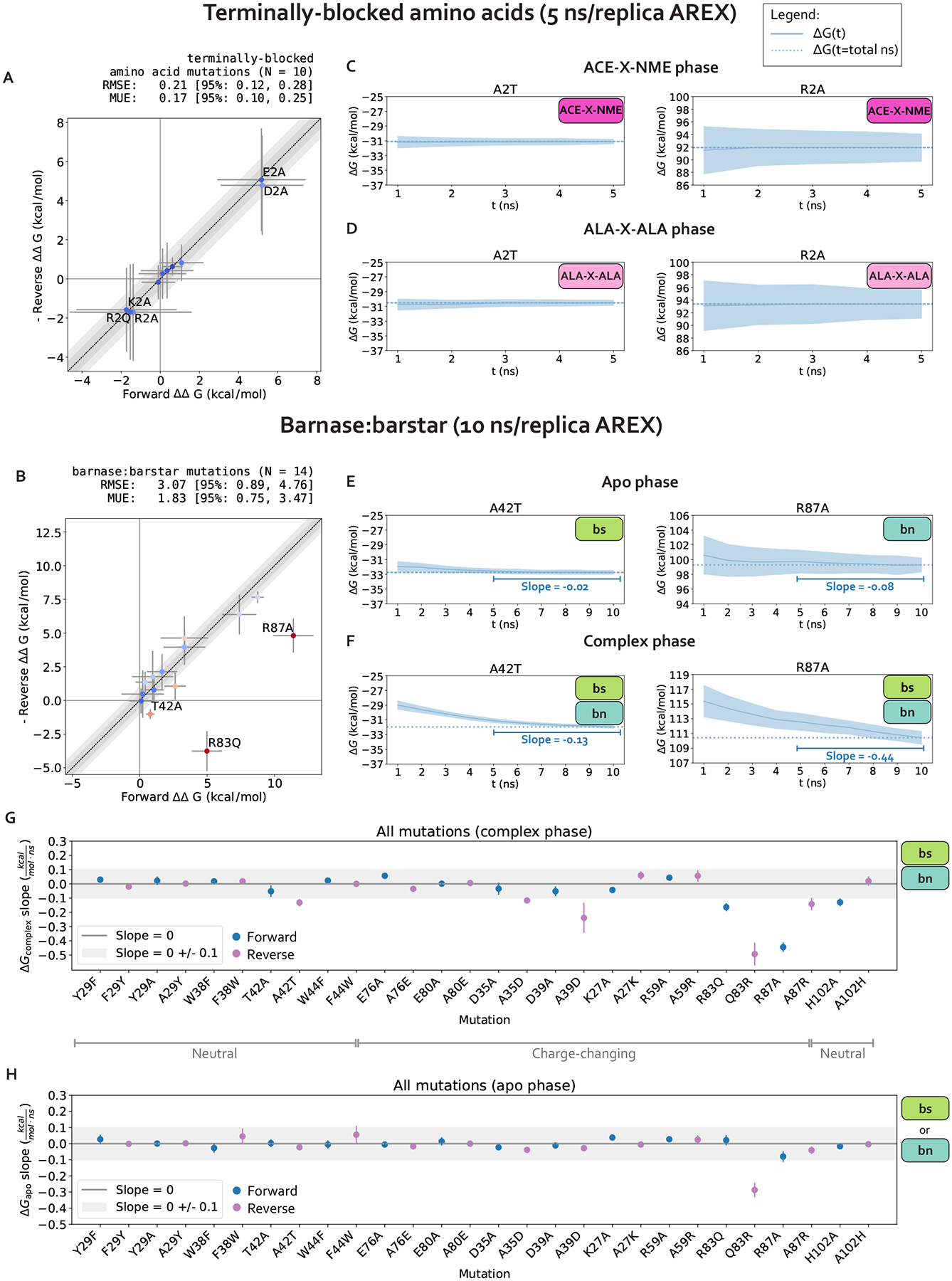
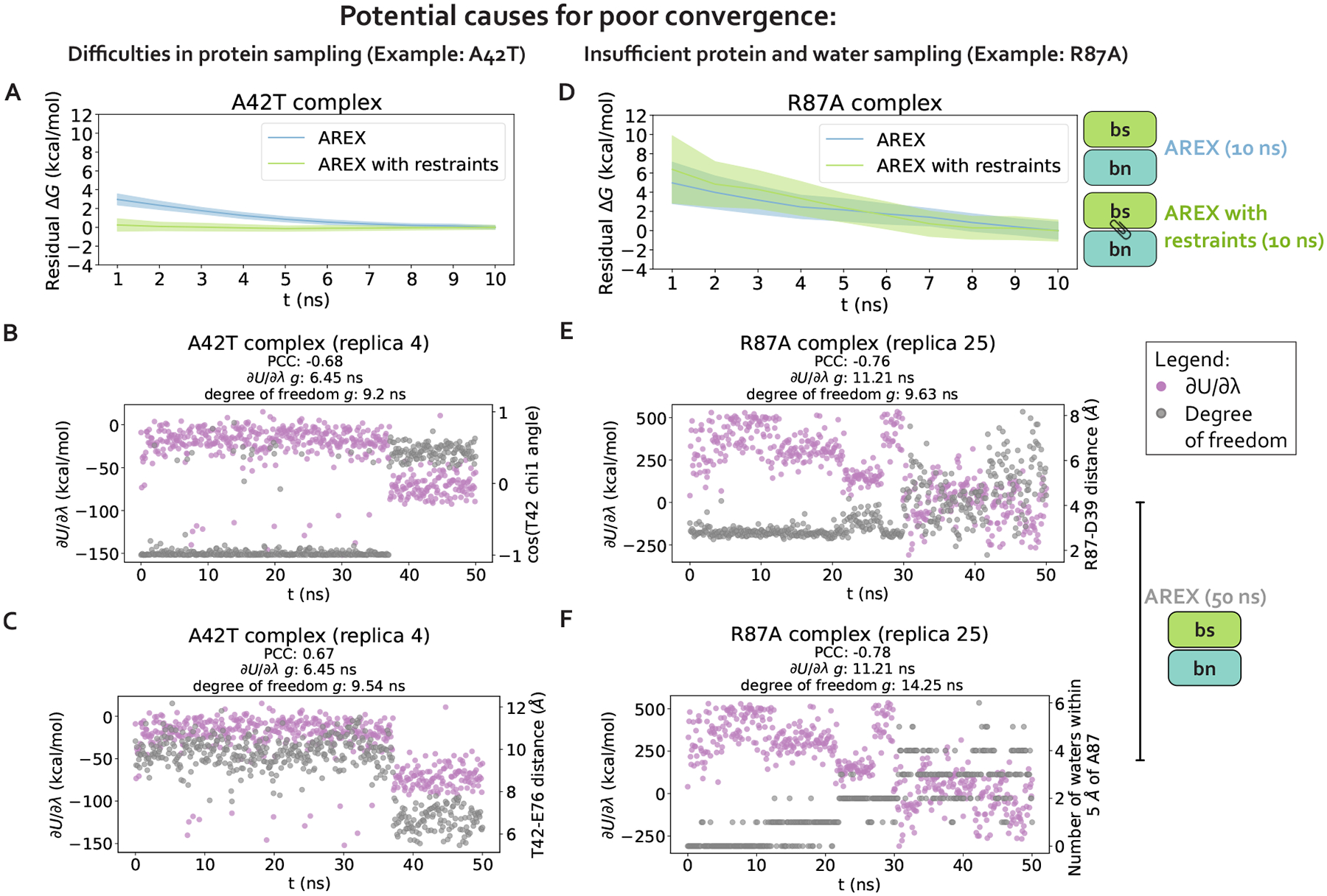
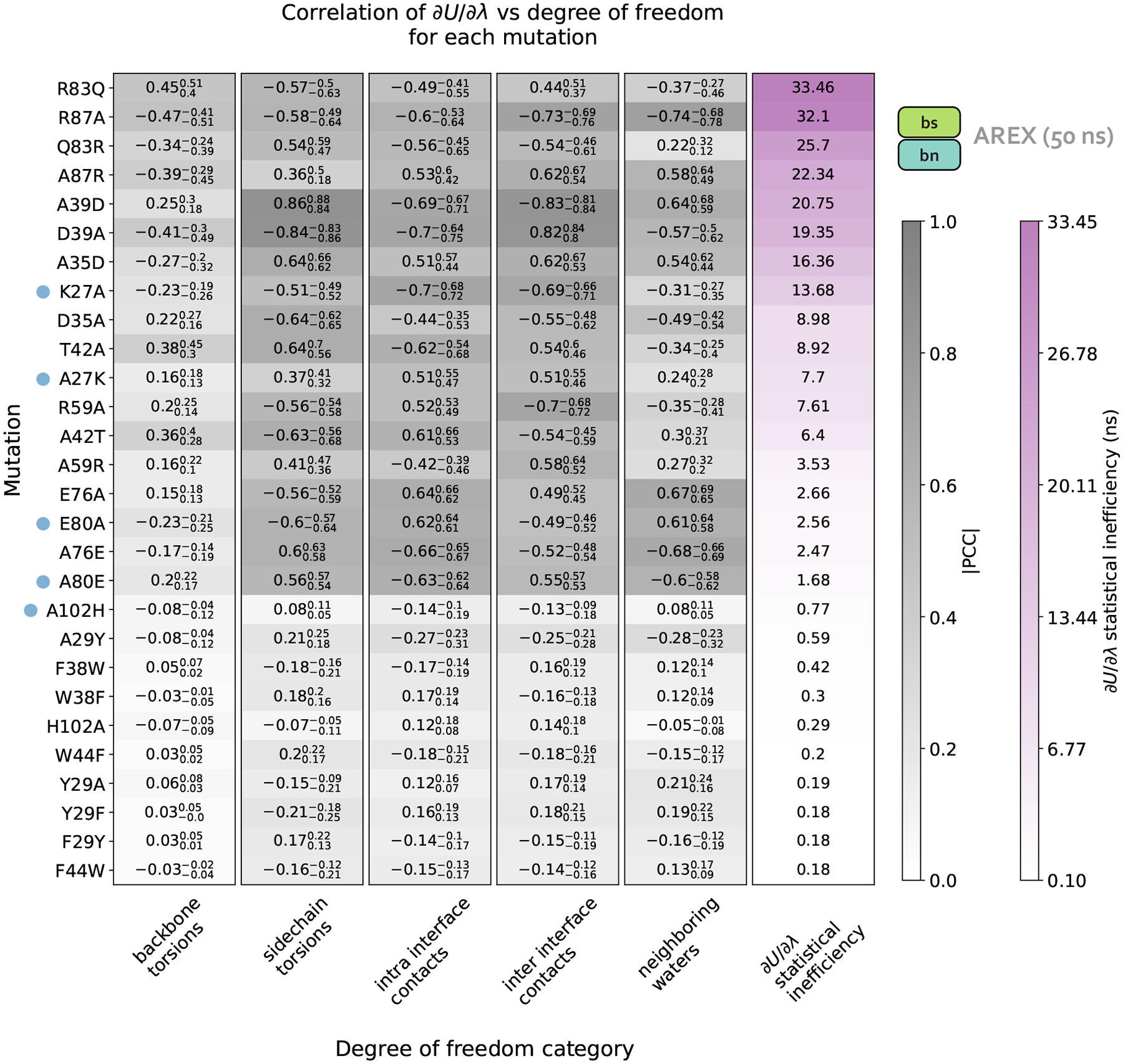
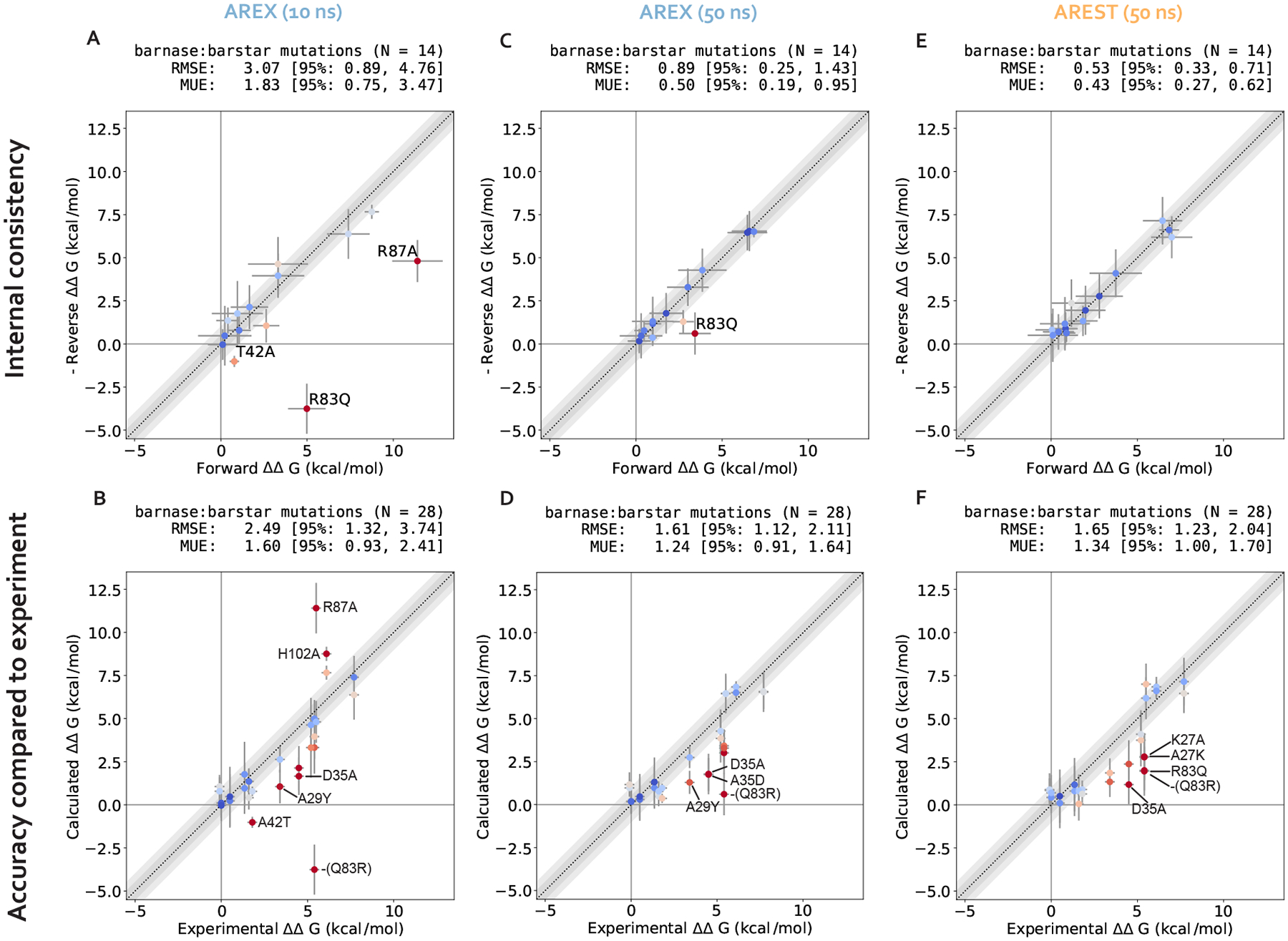
Relative alchemical binding free energy calculations are routinely used in drug discovery projects to optimize the affinity of small molecules for their drug targets. Alchemical methods can also be used to estimate the impact of amino acid mutations on protein:protein binding affinities, but these calculations can involve sampling challenges due to the complex networks of protein and water interactions frequently present in protein:protein interfaces. We investigate these challenges by extending a graphics processing unit (GPU)-accelerated open-source relative free energy calculation package (Perses) to predict the impact of amino acid mutations on protein:protein binding. Using the well-characterized model system barnase:barstar, we describe analyses for identifying and characterizing sampling problems in protein:protein relative free energy calculations. We find that mutations with sampling problems often involve charge-changes, and inadequate sampling can be attributed to slow degrees of freedom that are mutation-specific. We also explore the accuracy and efficiency of current state-of-the-art approaches─alchemical replica exchange and alchemical replica exchange with solute tempering─for overcoming relevant sampling problems. By employing sufficiently long simulations, we achieve accurate predictions (RMSE 1.61, 95% CI: [1.12, 2.11] kcal/mol), with 86% of estimates within 1 kcal/mol of the experimentally determined relative binding free energies and 100% of predictions correctly classifying the sign of the changes in binding free energies. Ultimately, we provide a model workflow for applying protein mutation free energy calculations to protein:protein complexes, and importantly, catalog the sampling challenges associated with these types of alchemical transformations. Our free open-source package (Perses) is based on OpenMM and is available at https://github.com/choderalab/perses.
相对结合自由能计算在药物发现项目中被广泛用于优化小分子与药物靶点的亲和力。 基于热力学积分的方法也可用于估计氨基酸突变对蛋白质-蛋白质结合亲和力的影响,但这些计算可能会涉及采样挑战,因为蛋白质-蛋白质界面中经常存在复杂的蛋白质和水相互作用网络。 我们通过将图形处理单元 (GPU) 加速的开源相对自由能计算包 (Perses) 扩展到预测氨基酸突变对蛋白质-蛋白质结合的影响来研究这些挑战。 我们使用经过充分表征的 barnase:barstar 模型系统,描述了用于识别和表征蛋白质-蛋白质相对自由能计算中采样问题的分析方法。 我们发现,具有采样问题的突变通常涉及电荷变化,并且采样不足可归因于特定突变的自由度较慢。 我们还探索了当前最先进的方法——基于热力学积分的 replica 交换和带溶剂温度调整的 replica 交换——克服相关采样问题的准确性和效率。 通过进行足够长的模拟,我们实现了准确的预测(RMSE 为 1.61,95%置信区间:[1.12, 2.11] kcal/mol),其中 86%的估计值与实验测定的相对结合自由能在 1 kcal/mol 以内,100%的预测值正确分类了结合自由能变化的符号。 最终,我们提供了一种应用蛋白质突变自由能计算到蛋白质-蛋白质复合物的模型工作流程,并重要的是,编目了与这些类型的热力学积分转换相关的采样挑战。 我们的免费开源软件包 (Perses) 基于 OpenMM,并可在 https://github.com/choderalab/perses 上获得。