Zhao Yingjie, Wang Yujue, Shi Lijie, McDonald-McGinn Donna M, Crowley T Blaine, McGinn Daniel E, Tran Oanh T, Miller Daniella, Lin Jhih-Rong, Zackai Elaine, Johnston H Richard, Chow Eva W C, Vorstman Jacob A S, Vingerhoets Claudia, van Amelsvoort Therese, Gothelf Doron, Swillen Ann, Breckpot Jeroen, Vermeesch Joris R, Eliez Stephan, Schneider Maude, van den Bree Marianne B M, Owen Michael J, Kates Wendy R, Repetto Gabriela M, Shashi Vandana, Schoch Kelly, Bearden Carrie E, Digilio M Cristina, Unolt Marta, Putotto Carolina, Marino Bruno, Pontillo Maria, Armando Marco, Vicari Stefano, Angkustsiri Kathleen, Campbell Linda, Busa Tiffany, Heine-Suñer Damian, Murphy Kieran C, Murphy Declan, García-Miñaúr Sixto, Fernández Luis, Zhang Zhengdong D, Goldmuntz Elizabeth, Gur Raquel E, Emanuel Beverly S, Zheng Deyou, Marshall Christian R, Bassett Anne S, Wang Tao, Morrow Bernice E
Department of Genetics, Albert Einstein College of Medicine, Bronx, NY, 10461, USA.
Division of Human Genetics, Children's Hospital of Philadelphia, Philadelphia, 19104, USA.
NPJ Genom Med. 2023 Jul 18;8(1):17. doi: 10.1038/s41525-023-00363-y.
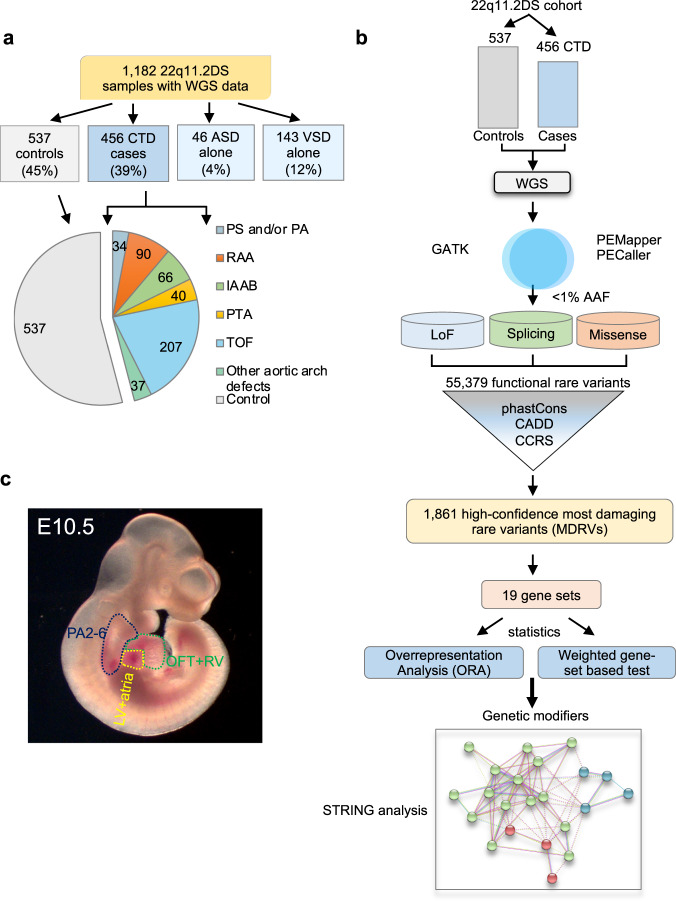
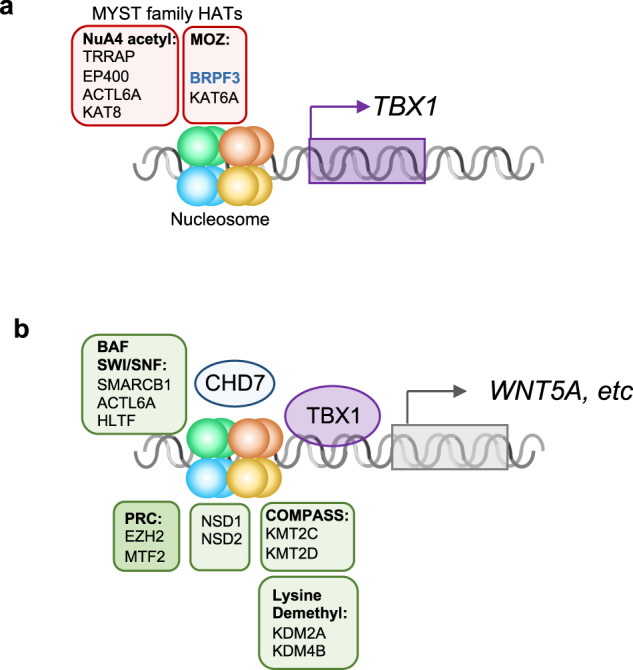
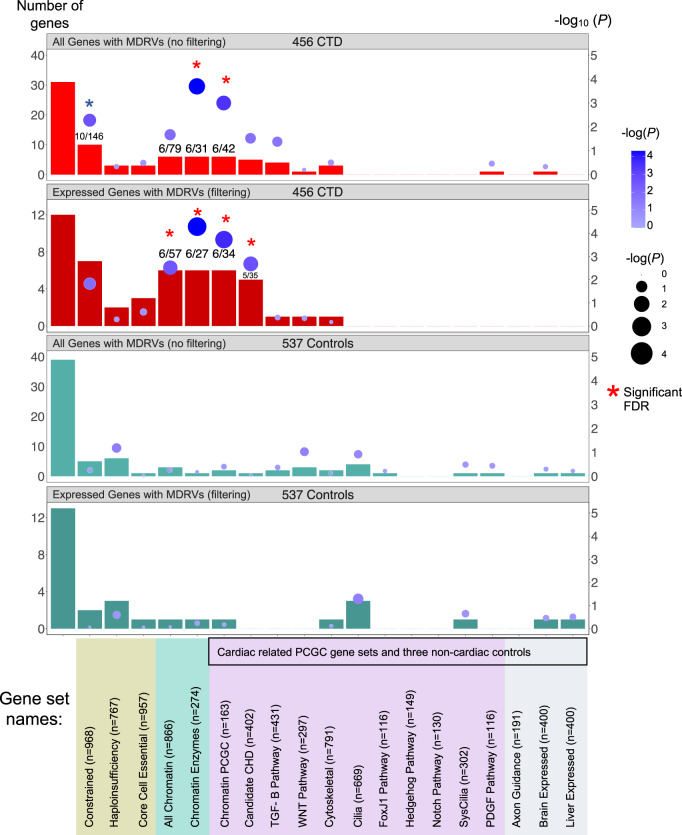
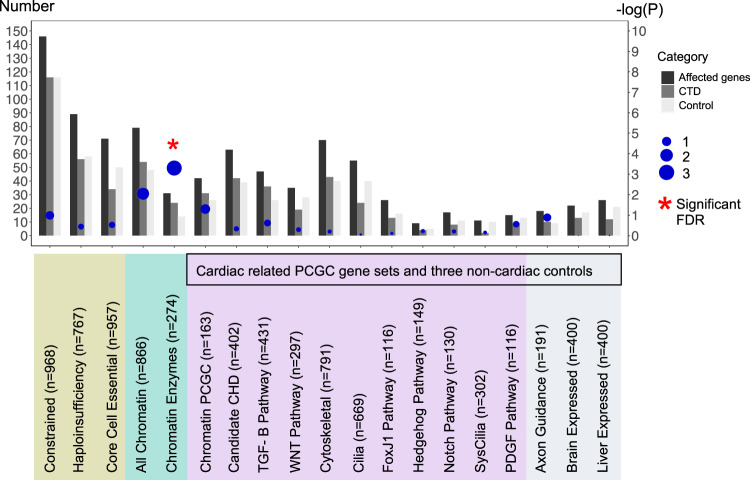
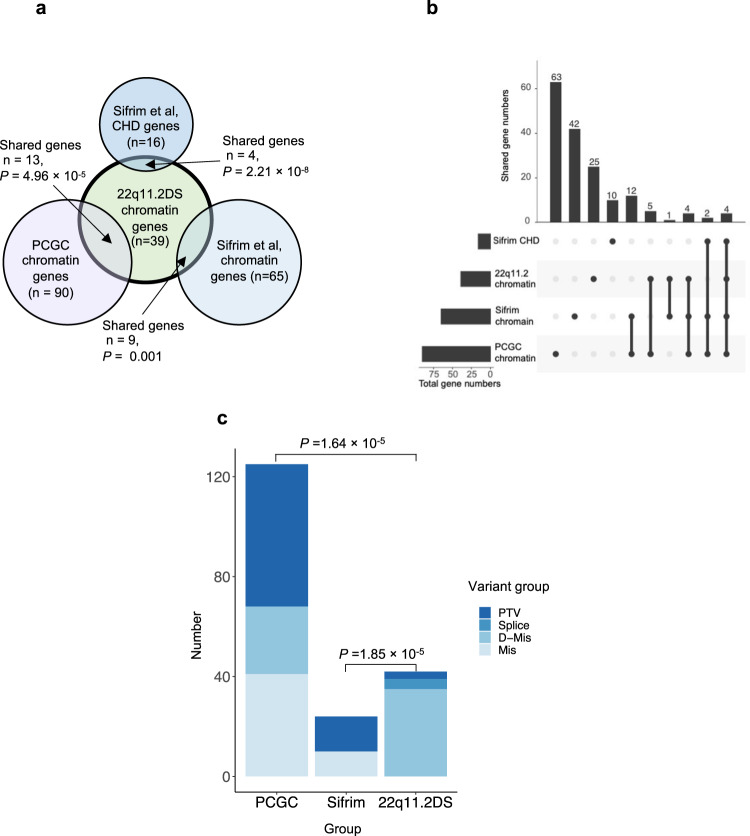
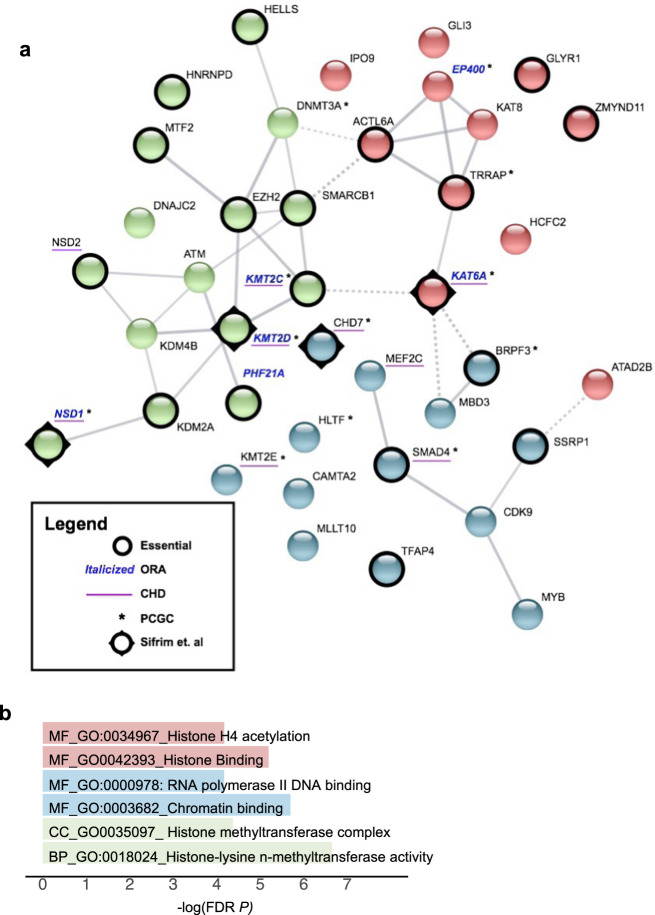
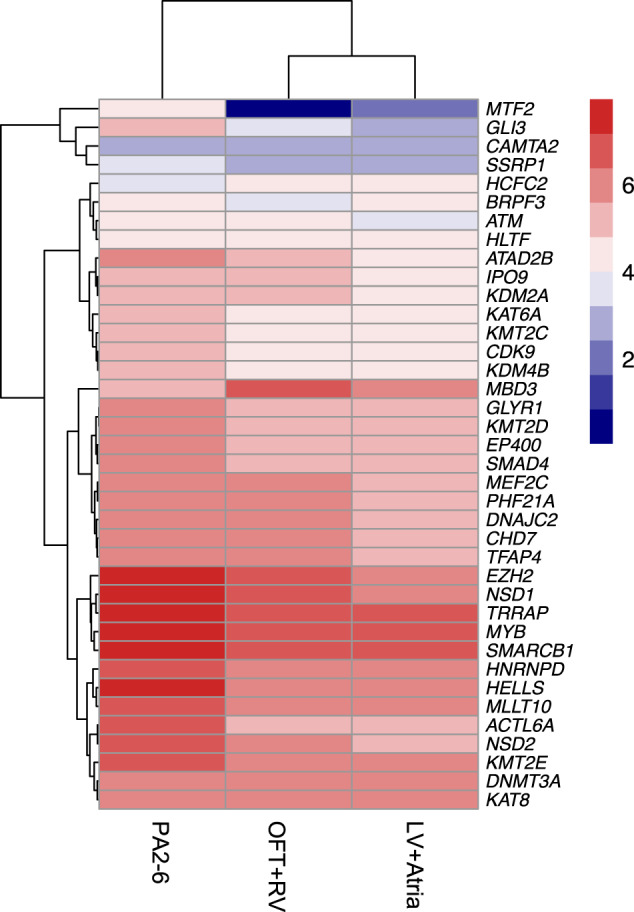
Congenital heart disease (CHD) affecting the conotruncal region of the heart, occurs in 40-50% of patients with 22q11.2 deletion syndrome (22q11.2DS). This syndrome is a rare disorder with relative genetic homogeneity that can facilitate identification of genetic modifiers. Haploinsufficiency of TBX1, encoding a T-box transcription factor, is one of the main genes responsible for the etiology of the syndrome. We suggest that genetic modifiers of conotruncal defects in patients with 22q11.2DS may be in the TBX1 gene network. To identify genetic modifiers, we analyzed rare, predicted damaging variants in whole genome sequence of 456 cases with conotruncal defects and 537 controls, with 22q11.2DS. We then performed gene set approaches and identified chromatin regulatory genes as modifiers. Chromatin genes with recurrent damaging variants include EP400, KAT6A, KMT2C, KMT2D, NSD1, CHD7 and PHF21A. In total, we identified 37 chromatin regulatory genes, that may increase risk for conotruncal heart defects in 8.5% of 22q11.2DS cases. Many of these genes were identified as risk factors for sporadic CHD in the general population. These genes are co-expressed in cardiac progenitor cells with TBX1, suggesting that they may be in the same genetic network. The genes KAT6A, KMT2C, CHD7 and EZH2, have been previously shown to genetically interact with TBX1 in mouse models. Our findings indicate that disturbance of chromatin regulatory genes impact the TBX1 gene network serving as genetic modifiers of 22q11.2DS and sporadic CHD, suggesting that there are some shared mechanisms involving the TBX1 gene network in the etiology of CHD.
先天性心脏病(CHD)累及心脏的圆锥动脉干区域,在22q11.2缺失综合征(22q11.2DS)患者中发生率为40%-50%。该综合征是一种罕见疾病,具有相对的基因同质性,这有助于识别基因修饰因子。编码T-box转录因子的TBX1单倍剂量不足是该综合征病因的主要基因之一。我们认为,22q11.2DS患者圆锥动脉干缺陷的基因修饰因子可能存在于TBX1基因网络中。为了识别基因修饰因子,我们分析了456例圆锥动脉干缺陷病例和537例22q11.2DS对照的全基因组序列中的罕见、预测有害变异。然后我们进行了基因集分析,并将染色质调节基因鉴定为修饰因子。具有复发性有害变异的染色质基因包括EP400、KAT6A、KMT2C、KMT2D、NSD1、CHD7和PHF21A。我们总共鉴定出37个染色质调节基因,它们可能使8.5%的22q11.2DS病例发生圆锥动脉干心脏缺陷的风险增加。这些基因中有许多被确定为普通人群散发性CHD的危险因素。这些基因在心脏祖细胞中与TBX1共表达,表明它们可能处于同一个基因网络中。基因KAT6A、KMT2C、CHD7和EZH2先前已在小鼠模型中显示与TBX1存在遗传相互作用。我们的研究结果表明,染色质调节基因的紊乱影响作为22q11.2DS和散发性CHD基因修饰因子的TBX1基因网络,这表明在CHD病因中存在一些涉及TBX1基因网络的共同机制。