Institute of Genomics, School of Medicine, Huaqiao University, 668 Jimei Road, Xiamen, 361021, China.
State Key Laboratory of Cellular Stress Biology, School of Life Sciences, Xiamen University, Xiamen, 361102, China.
BMC Biol. 2023 Jul 19;21(1):160. doi: 10.1186/s12915-023-01651-w.
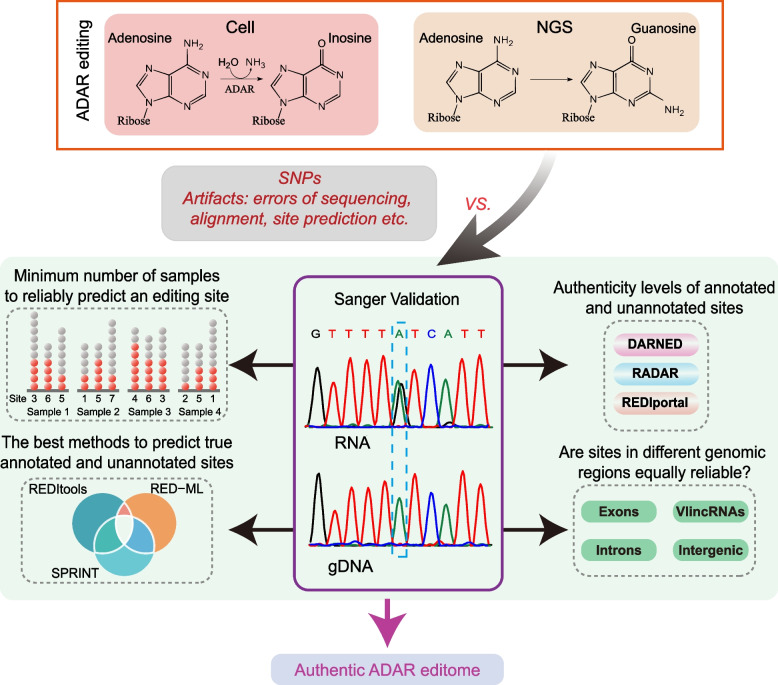
Conversion or editing of adenosine (A) into inosine (I) catalyzed by specialized cellular enzymes represents one of the most common post-transcriptional RNA modifications with emerging connection to disease. A-to-I conversions can happen at specific sites and lead to increase in proteome diversity and changes in RNA stability, splicing, and regulation. Such sites can be detected as adenine-to-guanine sequence changes by next-generation RNA sequencing which resulted in millions reported sites from multiple genome-wide surveys. Nonetheless, the lack of extensive independent validation in such endeavors, which is critical considering the relatively high error rate of next-generation sequencing, leads to lingering questions about the validity of the current compendiums of the editing sites and conclusions based on them.
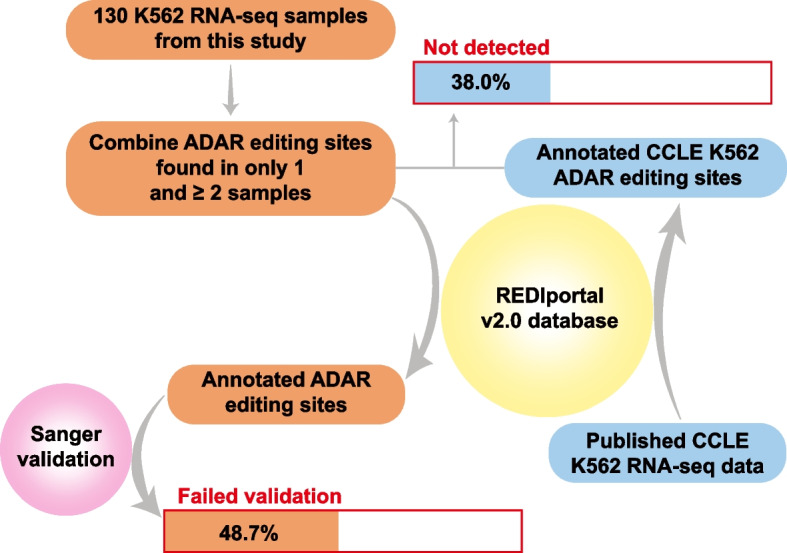
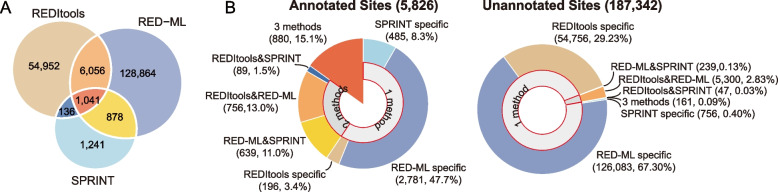
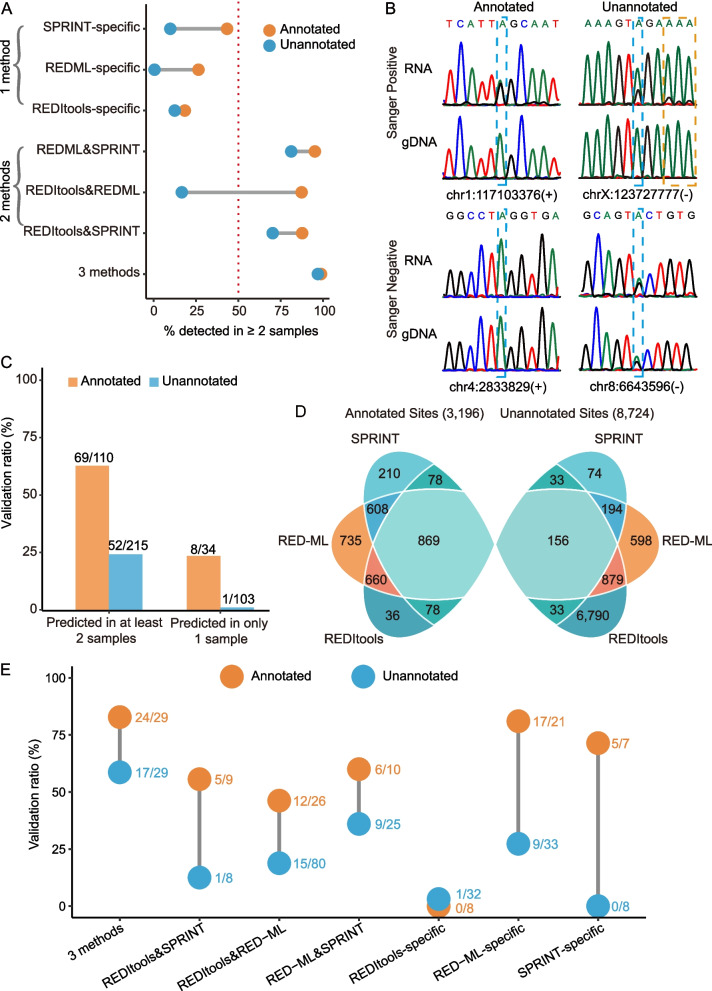
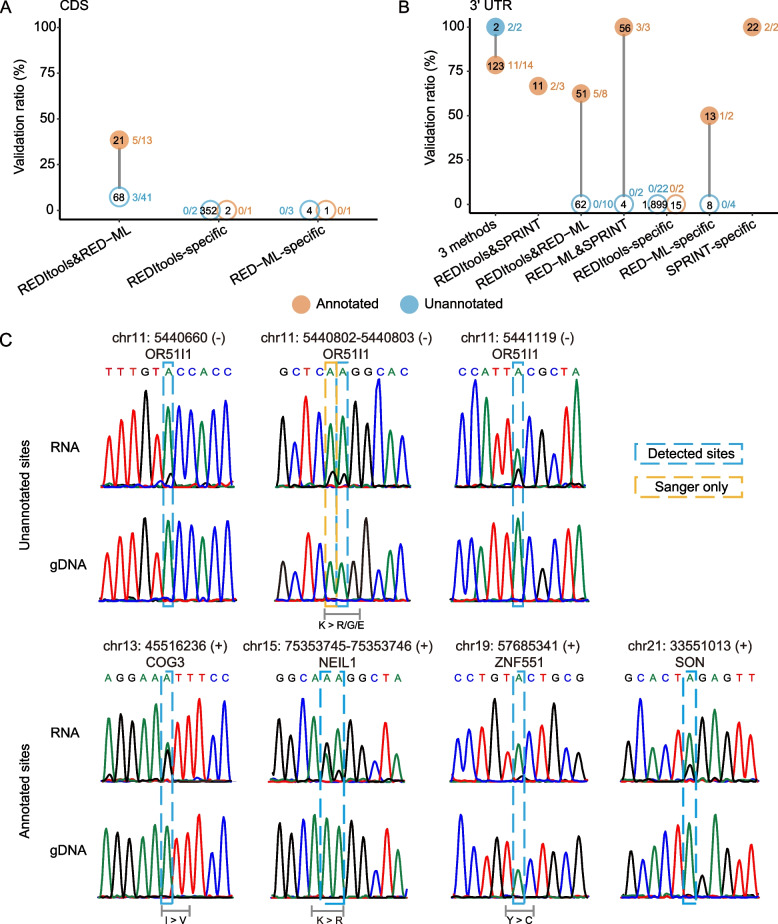
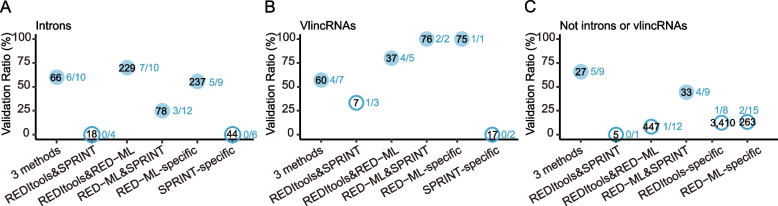
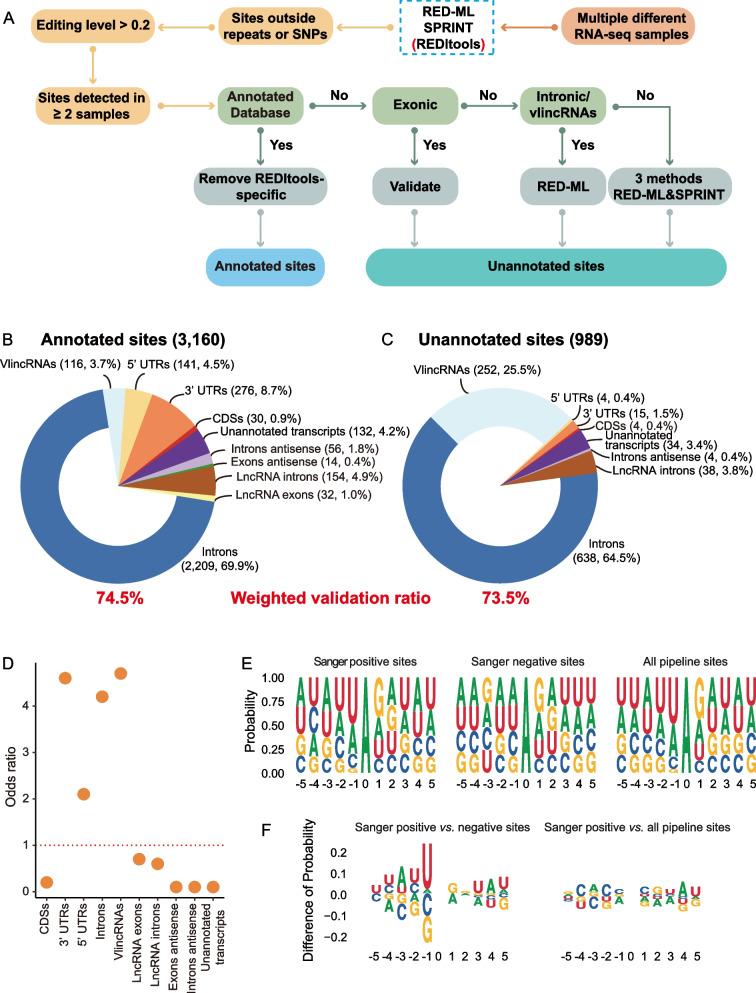
Strikingly, we found that the current analytical methods suffer from very high false positive rates and that a significant fraction of sites in the public databases cannot be validated. In this work, we present potential solutions to these problems and provide a comprehensive and extensively validated list of A-to-I editing sites in a human cancer cell line. Our findings demonstrate that most of true A-to-I editing sites in a human cancer cell line are located in the non-coding transcripts, the so-called RNA 'dark matter'. On the other hand, many ADAR editing events occurring in exons of human protein-coding mRNAs, including those that can recode the transcriptome, represent false positives and need to be interpreted with caution. Nonetheless, yet undiscovered authentic ADAR sites that increase the diversity of human proteome exist and warrant further identification.
Accurate identification of human ADAR sites remains a challenging problem, particularly for the sites in exons of protein-coding mRNAs. As a result, genome-wide surveys of ADAR editome must still be accompanied by extensive Sanger validation efforts. However, given the vast number of unknown human ADAR sites, there is a need for further developments of the analytical techniques, potentially those that are based on deep learning solutions, in order to provide a quick and reliable identification of the editome in any sample.
由专门的细胞酶催化的腺苷(A)转化或编辑为肌苷(I)是最常见的转录后 RNA 修饰之一,与疾病的关系正逐渐被揭示。A 到 I 的转换可以在特定位置发生,并导致蛋白质组多样性增加,以及 RNA 稳定性、剪接和调控的变化。通过下一代 RNA 测序可以检测到这些位点的腺嘌呤到鸟嘌呤序列变化,这导致从多个全基因组调查中报告了数百万个位点。然而,在这些研究中缺乏广泛的独立验证,考虑到下一代测序的相对较高错误率,这对于当前编辑位点的汇编以及基于这些汇编的结论的有效性仍然存在疑问。
令人惊讶的是,我们发现当前的分析方法存在非常高的假阳性率,并且公共数据库中的许多位点无法验证。在这项工作中,我们提出了这些问题的潜在解决方案,并提供了人类癌细胞系中 A 到 I 编辑位点的全面和广泛验证的列表。我们的研究结果表明,人类癌细胞系中大多数真正的 A 到 I 编辑位点位于非编码转录本中,即所谓的 RNA“暗物质”。另一方面,许多发生在人类蛋白质编码 mRNA 外显子中的 ADAR 编辑事件,包括那些可以重新编码转录组的事件,都是假阳性,需要谨慎解释。然而,仍然存在增加人类蛋白质组多样性的未被发现的真实 ADAR 位点,需要进一步鉴定。
准确识别人类 ADAR 位点仍然是一个具有挑战性的问题,特别是对于蛋白质编码 mRNA 外显子中的位点。因此,ADAR 编辑组的全基因组调查仍然需要与广泛的 Sanger 验证工作相结合。然而,鉴于大量未知的人类 ADAR 位点,需要进一步开发分析技术,可能是基于深度学习解决方案的技术,以便在任何样本中快速可靠地识别编辑组。