Department of Chemistry, Faculty of Sciences, Persian Gulf University, Bushehr, 75169, Iran.
Stem Cells Technology Research Center, Shiraz University of Medical Sciences, Shiraz, Iran.
Sci Rep. 2023 Jul 24;13(1):11952. doi: 10.1038/s41598-023-38236-0.
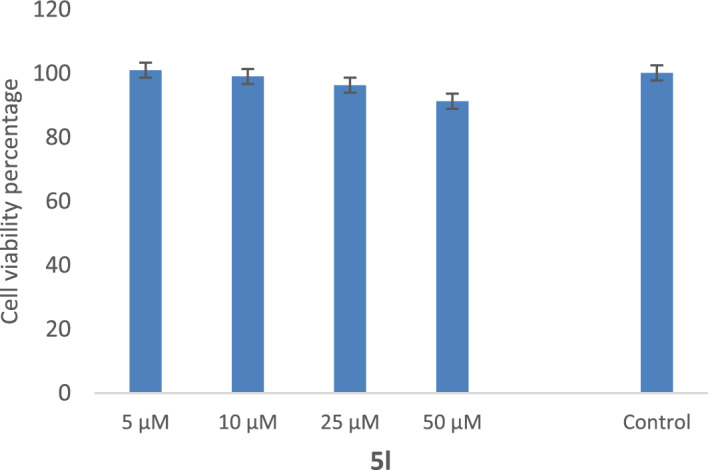
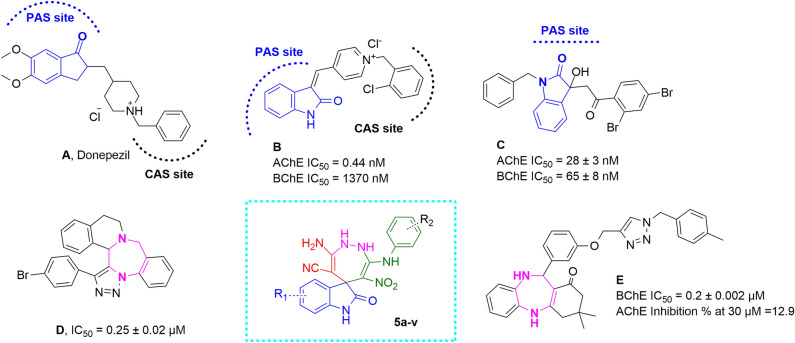
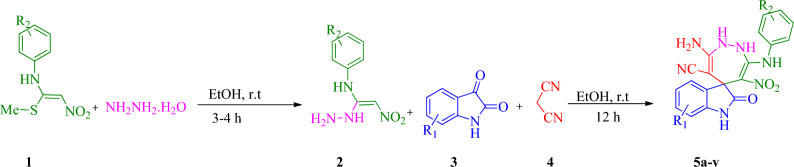
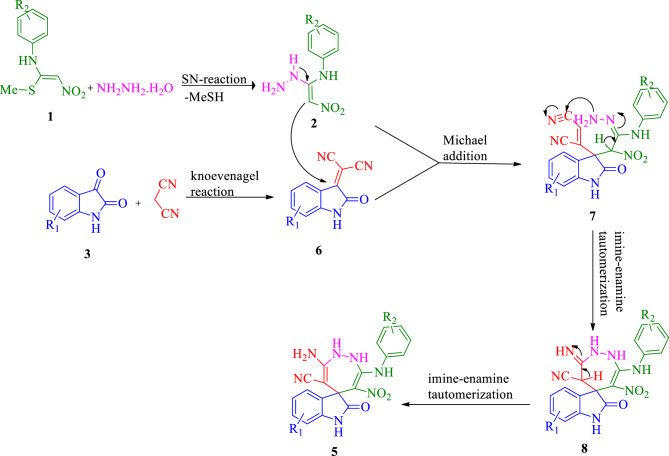
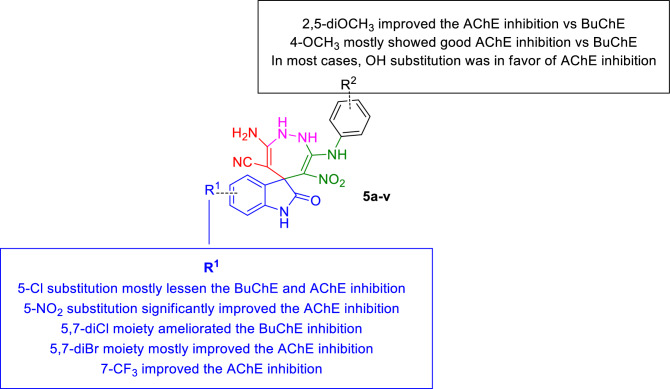
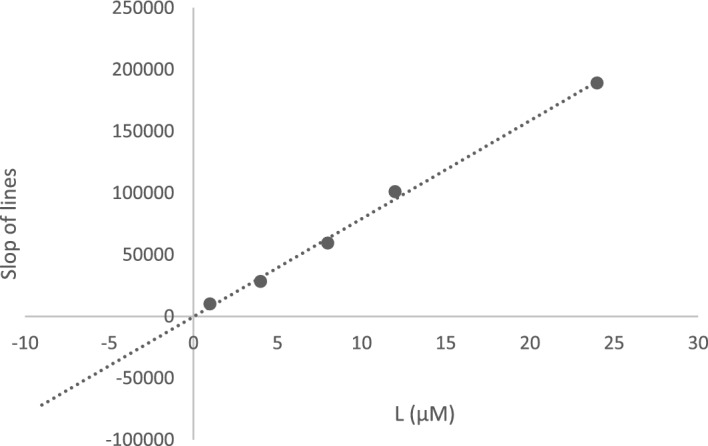
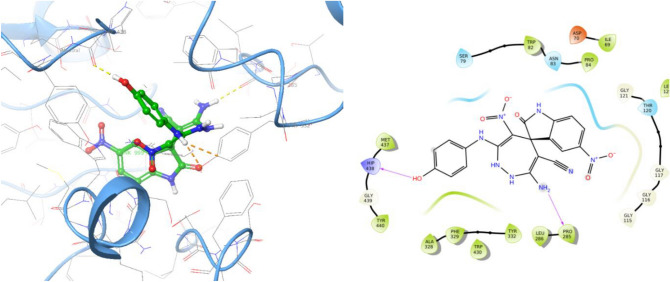
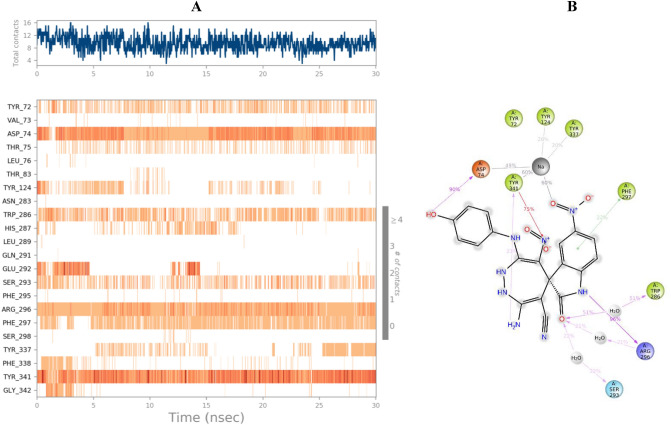
In this study, a new series of spiro indolin-1,2-diazepine were designed, synthesized, and screened for their cholinesterase inhibitory activities. A novel, green, high-yielding approach was constructed to synthesize spiro indolin-1,2-diazepine derivatives through a cascade reaction of different isatins, malononitrile and 1,1-enediamines (EDAMs) via sequential four-component reactions to produce the target compounds with good to excellent yields. Next the inhibitory potencies of all derivatives were determined spectroscopically at 415 nm using the modified Ellman method. The results of the in vitro screening indicated that 5l with spiroindolin-1,2-diazepine core bearing 5-NO at R and 4-OH at R was the most potent and selective AChE inhibitor with an IC value of 3.98 ± 1.07 µM with no significant inhibition against BChE while 5j was the most active analog against both AChE and BChE enzymes. The structure-activity relationships suggested the variation in the inhibitory activities of derivatives was affected by different substitutions on the indolinone ring as well as the phenyl moiety. The enzyme kinetic studies of the most potent compound 5l at five different concentrations and acetylthiocholine substrate (0.1-1 mM) by Ellman's method revealed that it inhibited AChE in a mixed mode with a K of 0.044 μM. A molecular docking study was performed via induced fit docking protocol to predict the putative binding interaction. It was shown that the moieties used in the initial structure design play a fundamental role in interacting with the enzyme's binding site. Further, molecular dynamics simulations with the Schrödinger package were performed for 5l in a complex with AChE and revealed that compound 5l formed the stable complex with the enzyme. The MTT toxicity assessments against the neuroblastoma cell line were executed, and no toxicity was seen for 5l under the tested concentrations.
在这项研究中,设计、合成了一系列新的螺环吲哚啉-1,2-二氮杂卓,并对其胆碱酯酶抑制活性进行了筛选。通过不同的靛红、丙二腈和 1,1-二乙二胺(EDAMs)的级联反应,构建了一种新颖的、绿色的、高产的方法来合成螺环吲哚啉-1,2-二氮杂卓衍生物,通过连续的四组分反应以良好到优异的产率生成目标化合物。接下来,通过改良的 Ellman 法在 415nm 处光谱测定所有衍生物的抑制活力。体外筛选结果表明,R 位带有 5-NO 和 R 位带有 4-OH 的螺环吲哚啉-1,2-二氮杂卓核心的 5l 对 AChE 的抑制作用最强且最具选择性,IC 值为 3.98±1.07µM,对 BChE 无明显抑制作用,而 5j 是对 AChE 和 BChE 酶最具活性的类似物。构效关系表明,取代基的变化会影响衍生物的抑制活性,包括吲哚酮环和苯环部分。通过 Ellman 法在五个不同浓度下和乙酰硫代胆碱底物(0.1-1mM)对最有效的化合物 5l 进行酶动力学研究,结果表明它以混合模式抑制 AChE,K 值为 0.044µM。通过诱导拟合对接方案进行了分子对接研究,以预测可能的结合相互作用。结果表明,在初始结构设计中使用的部分在与酶的结合位点相互作用中起着基本作用。此外,还使用 Schrödinger 包对 5l 与 AChE 的复合物进行了分子动力学模拟,结果表明化合物 5l 与酶形成了稳定的复合物。对神经母细胞瘤细胞系进行了 MTT 毒性评估,在测试浓度下 5l 没有显示出毒性。