van Vlimmeren Anne E, Voleti Rashmi, Chartier Cassandra A, Jiang Ziyuan, Karandur Deepti, Humphries Preston A, Lo Wan-Lin, Shah Neel H
Department of Chemistry, Columbia University, New York, NY 10027.
Department of Biological Sciences, Columbia University, New York, NY 10027.
bioRxiv. 2024 Apr 9:2023.07.10.548257. doi: 10.1101/2023.07.10.548257.
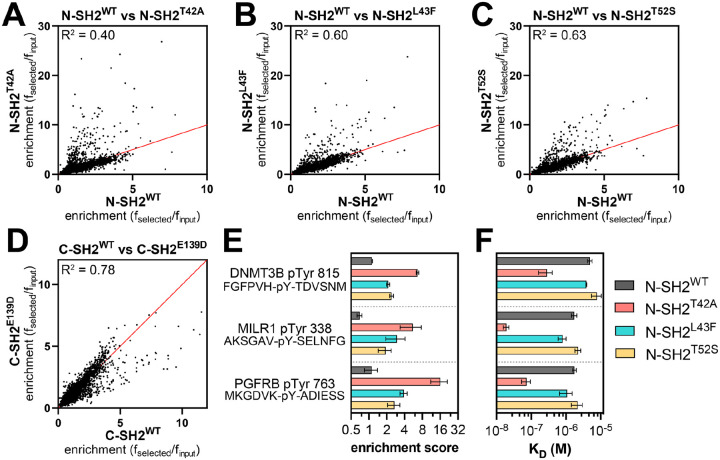
Mutations in the tyrosine phosphatase SHP2 are associated with a variety of human diseases. Most mutations in SHP2 increase its basal catalytic activity by disrupting auto-inhibitory interactions between its phosphatase domain and N-terminal SH2 (phosphotyrosine recognition) domain. By contrast, some disease-associated mutations located in the ligand-binding pockets of the N- or C-terminal SH2 domains do not increase basal activity and likely exert their pathogenicity through alternative mechanisms. We lack a molecular understanding of how these SH2 mutations impact SHP2 structure, activity, and signaling. Here, we characterize five SHP2 SH2 domain ligand-binding pocket mutants through a combination of high-throughput biochemical screens, biophysical and biochemical measurements, and molecular dynamics simulations. We show that, while some of these mutations alter binding affinity to phosphorylation sites, the T42A mutation in the N-SH2 domain is unique in that it also substantially alters ligand-binding specificity, despite being 8-10 Å from the specificity-determining region of the SH2 domain. This mutation exerts its effect on sequence specificity by remodeling the phosphotyrosine binding pocket, altering the mode of engagement of both the phosphotyrosine and surrounding residues on the ligand. The functional consequence of this altered specificity is that the T42A mutant has biased sensitivity toward a subset of activating ligands and enhances downstream signaling. Our study highlights an example of a nuanced mechanism of action for a disease-associated mutation, characterized by a change in protein-protein interaction specificity that alters enzyme activation.
酪氨酸磷酸酶SHP2的突变与多种人类疾病相关。SHP2中的大多数突变通过破坏其磷酸酶结构域与N端SH2(磷酸酪氨酸识别)结构域之间的自抑制相互作用来增加其基础催化活性。相比之下,位于N端或C端SH2结构域配体结合口袋中的一些疾病相关突变不会增加基础活性,可能通过其他机制发挥其致病性。我们对这些SH2突变如何影响SHP2的结构、活性和信号传导缺乏分子层面的理解。在这里,我们通过高通量生化筛选、生物物理和生化测量以及分子动力学模拟相结合的方法,对五个SHP2 SH2结构域配体结合口袋突变体进行了表征。我们发现,虽然其中一些突变改变了对磷酸化位点的结合亲和力,但N-SH2结构域中的T42A突变是独特的,因为它也显著改变了配体结合特异性,尽管它距离SH2结构域的特异性决定区域有8-10埃。该突变通过重塑磷酸酪氨酸结合口袋、改变配体上磷酸酪氨酸和周围残基的结合模式来对序列特异性产生影响。这种特异性改变的功能后果是,T42A突变体对一部分激活配体具有偏向性敏感性,并增强下游信号传导。我们的研究突出了一个疾病相关突变的细微作用机制的例子,其特征是蛋白质-蛋白质相互作用特异性的改变会改变酶的激活。