Talkhatova Sandugash, Aripov Marat, Mussayev Abdurashid, Alimbayev Serik, Otarbayev Yerik, Pya Yuri
National Research Cardiac Surgery Center, Astana 010000, Kazakhstan.
Department of Interventional Cardiology, National Research Cardiac Surgery Center, Astana 010000, Kazakhstan.
Int J Surg Case Rep. 2023 Aug;109:108521. doi: 10.1016/j.ijscr.2023.108521. Epub 2023 Jul 21.
Anomalous origin of the left coronary artery from the pulmonary artery (ALCAPA) is a rare congenital heart disease that usually presents with heart failure symptoms in infants. Without surgical correction, the condition has a high infant mortality rate. However, patients with ALCAPA can remain asymptomatic for decades in some cases, and the risk of sudden death decreases in adulthood.
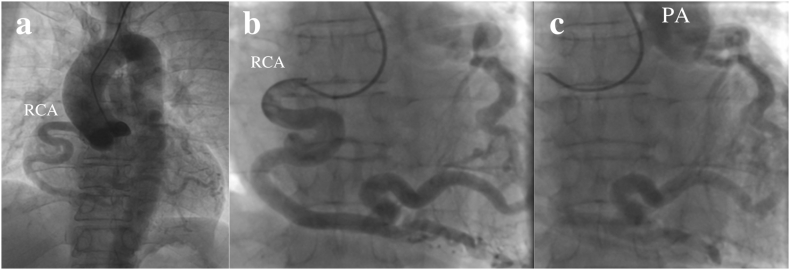
We present the case of a 52-year-old female who was incidentally diagnosed with ALCAPA during a routine medical evaluation. As the patient age, was asymptomatic, had good coronary collateral circulation, a medical treatment strategy was chosen and the patient was discharged in a good physical condition. And during the three-month follow-up, no cardiovascular complications were observed.
The appearance and severity of symptoms in patients with ALCAPA can vary depending on factors such as the closure of the patent ductus arteriosus (PDA), pressure gradient between arteries, collateral development, and coronary anatomy. Surgical intervention is typically recommended, but in select cases such us, conservative management may be considered for elderly patients due to increased surgical risks and potentially lower risk of sudden cardiac death. Individualized patient assessment is crucial in determining the optimal treatment strategy for ALCAPA, considering the available evidence and limitations.
The management of asymptomatic patients with ALCAPA remains a subject of discussion, and further research is needed to standardize the clinical approach for this subgroup of patients and to compare survival rates between surgical correction and medical therapy.
左冠状动脉起源于肺动脉(ALCAPA)是一种罕见的先天性心脏病,通常在婴儿期出现心力衰竭症状。未经手术矫正,该病婴儿死亡率很高。然而,在某些情况下,ALCAPA患者可能数十年无症状,成年后猝死风险降低。
我们报告一例52岁女性,在常规医学评估中偶然诊断为ALCAPA。由于患者年龄较大、无症状、冠状动脉侧支循环良好,因此选择了药物治疗策略,患者出院时身体状况良好。在三个月的随访中,未观察到心血管并发症。
ALCAPA患者症状的表现和严重程度可能因动脉导管未闭(PDA)的闭合、动脉之间的压力梯度、侧支循环发育和冠状动脉解剖结构等因素而有所不同。通常建议进行手术干预,但在某些特定情况下,如本例,由于手术风险增加且心脏性猝死风险可能较低,老年患者可考虑保守治疗。考虑到现有证据和局限性,个体化的患者评估对于确定ALCAPA的最佳治疗策略至关重要。
无症状ALCAPA患者的管理仍是一个讨论的话题,需要进一步研究以规范该亚组患者的临床方法,并比较手术矫正和药物治疗的生存率。