Robledo-Ruiz Diana A, Austin Lana, Amos J Nevil, Castrejón-Figueroa Jesús, Harley Daniel K P, Magrath Michael J L, Sunnucks Paul, Pavlova Alexandra
School of Biological Sciences, Monash University, Clayton, Victoria, Australia.
Department of Energy, Environment and Climate Action, Arthur Rylah Institute for Environmental Research, Heidelberg, Victoria, Australia.
Mol Ecol Resour. 2025 Jul;25(5):e13844. doi: 10.1111/1755-0998.13844. Epub 2023 Aug 1.
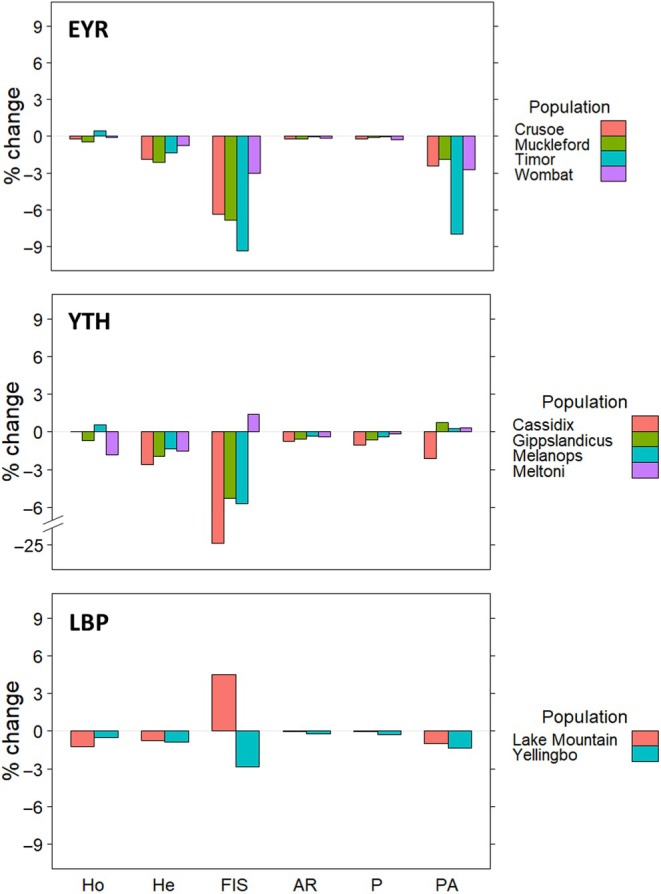
Identifying sex-linked markers in genomic datasets is important because their presence in supposedly neutral autosomal datasets can result in incorrect estimates of genetic diversity, population structure and parentage. However, detecting sex-linked loci can be challenging, and available scripts neglect some categories of sex-linked variation. Here, we present new R functions to (1) identify and separate sex-linked loci in ZW and XY sex determination systems and (2) infer the genetic sex of individuals based on these loci. We tested these functions on genomic data for two bird and one mammal species and compared the biological inferences made before and after removing sex-linked loci using our function. We found that our function identified autosomal loci with ≥98.8% accuracy and sex-linked loci with an average accuracy of 87.8%. We showed that standard filters, such as low read depth and call rate, failed to remove up to 54.7% of sex-linked loci. This led to (i) overestimation of population F by up to 24%, and the number of private alleles by up to 8%; (ii) wrongly inferring significant sex differences in heterozygosity; (iii) obscuring genetic population structure and (iv) inferring ~11% fewer correct parentages. We discuss how failure to remove sex-linked markers can lead to incorrect biological inferences (e.g. sex-biased dispersal and cryptic population structure) and misleading management recommendations. For reduced-representation datasets with at least 15 known-sex individuals of each sex, our functions offer convenient resources to remove sex-linked loci and to sex the remaining individuals (freely available at https://github.com/drobledoruiz/conservation_genomics).
在基因组数据集中识别性连锁标记很重要,因为它们出现在假定为中性的常染色体数据集中会导致对遗传多样性、种群结构和亲子关系的估计错误。然而,检测性连锁基因座可能具有挑战性,并且现有的脚本忽略了某些类型的性连锁变异。在这里,我们提出了新的R函数,用于(1)在ZW和XY性别决定系统中识别和分离性连锁基因座,以及(2)根据这些基因座推断个体的遗传性别。我们在两种鸟类和一种哺乳动物的基因组数据上测试了这些函数,并比较了使用我们的函数去除性连锁基因座前后所做的生物学推断。我们发现我们的函数识别常染色体基因座的准确率≥98.8%,识别性连锁基因座的平均准确率为87.8%。我们表明,标准过滤方法,如低读取深度和调用率,未能去除高达54.7%的性连锁基因座。这导致了:(i)种群F高估高达24%,私有等位基因数量高估高达8%;(ii)错误推断杂合性的显著性别差异;(iii)掩盖遗传种群结构;(iv)正确推断的亲子关系减少约11%。我们讨论了未能去除性连锁标记如何导致错误的生物学推断(例如性别偏向扩散和隐秘种群结构)以及误导性的管理建议。对于每种性别至少有15个已知性别的个体的简化代表性数据集,我们的函数提供了方便的资源来去除性连锁基因座并对其余个体进行性别鉴定(可在https://github.com/drobledoruiz/conservation_genomics免费获取)。