Turku Bioscience Centre, University of Turku and Åbo Akademi University, Turku, Finland.
InFLAMES Research Flagship Center, University of Turku, Turku, Finland.
PLoS Comput Biol. 2023 Aug 11;19(8):e1010727. doi: 10.1371/journal.pcbi.1010727. eCollection 2023 Aug.
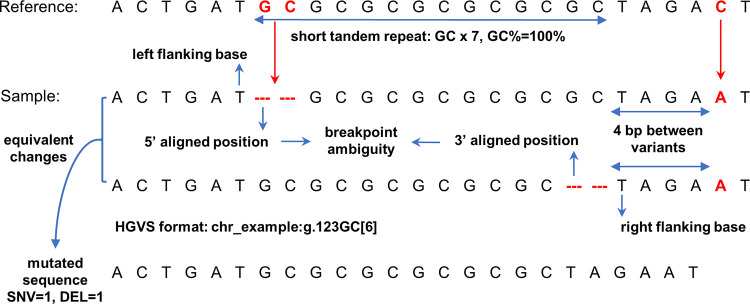
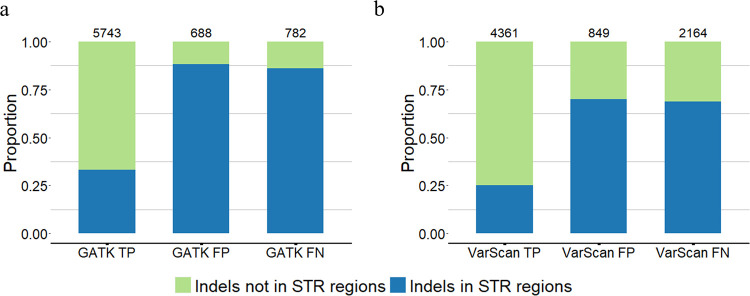
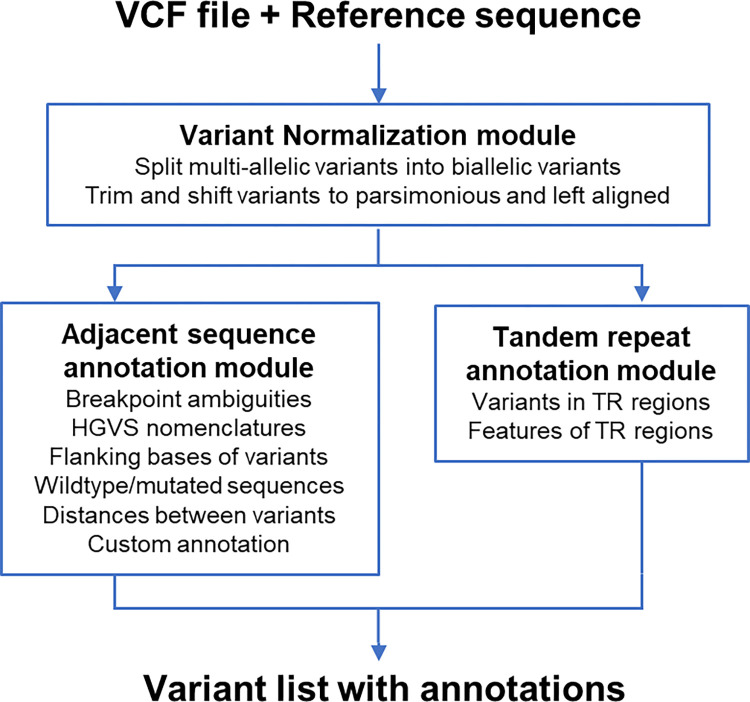
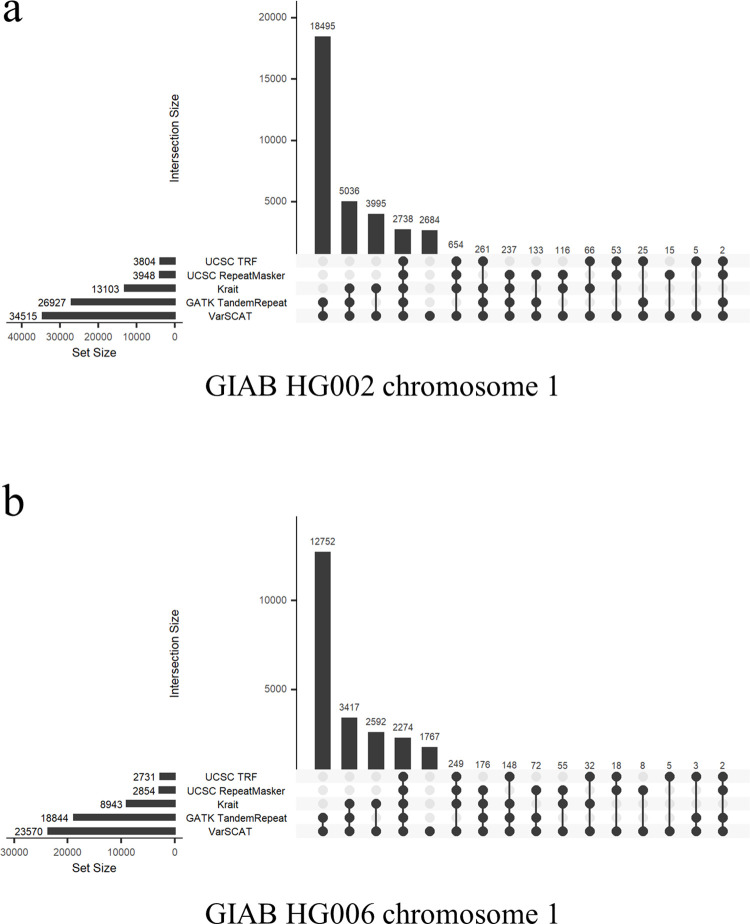
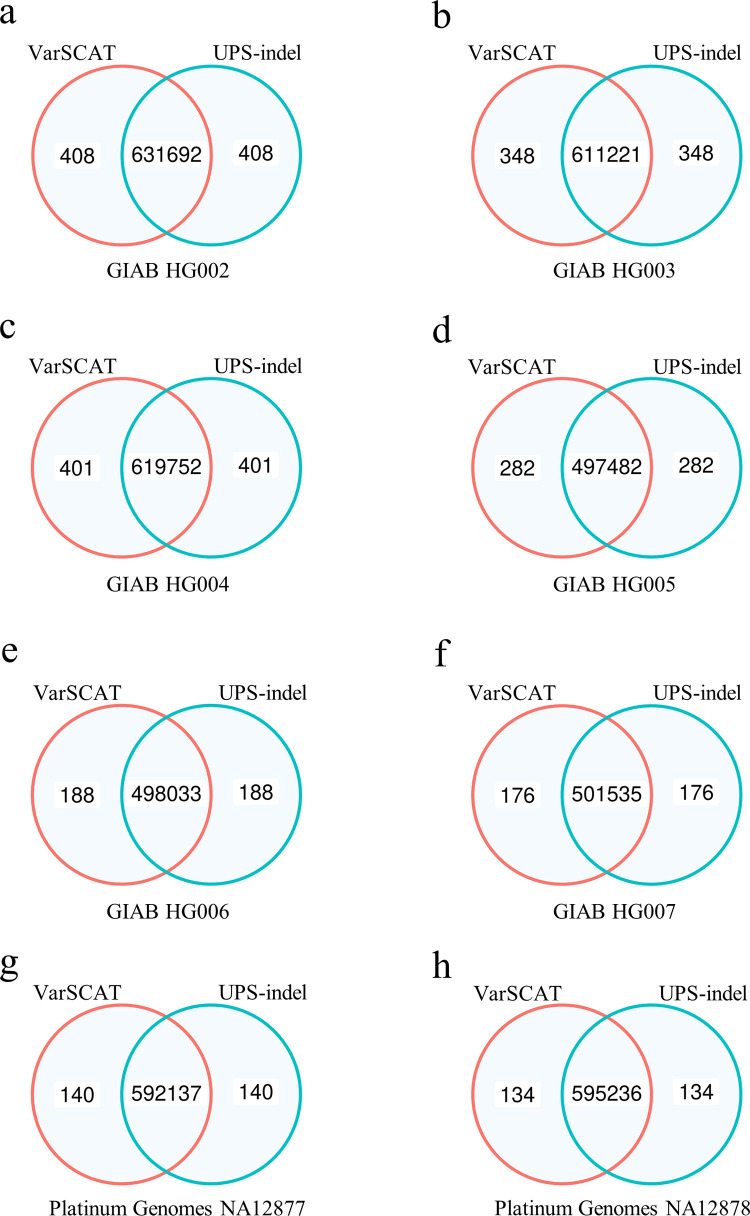
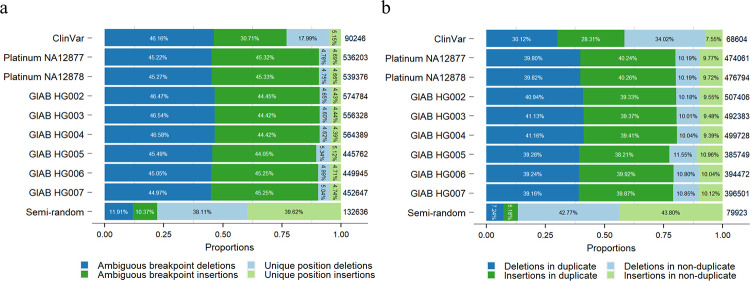
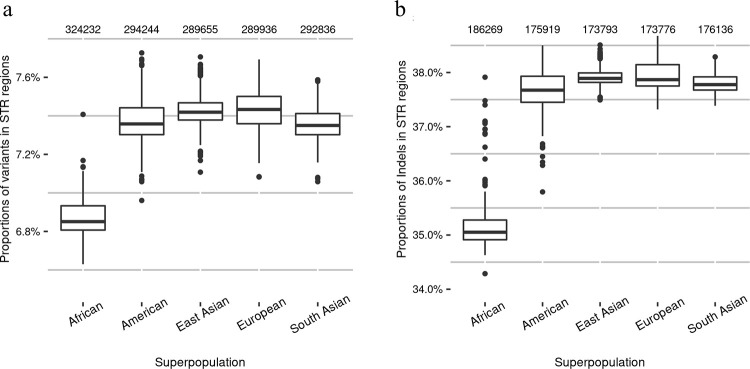
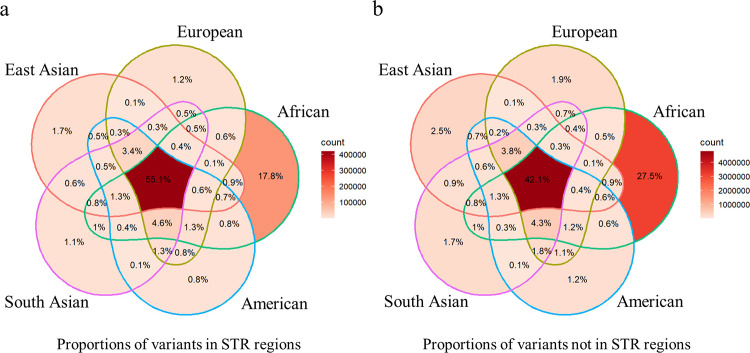
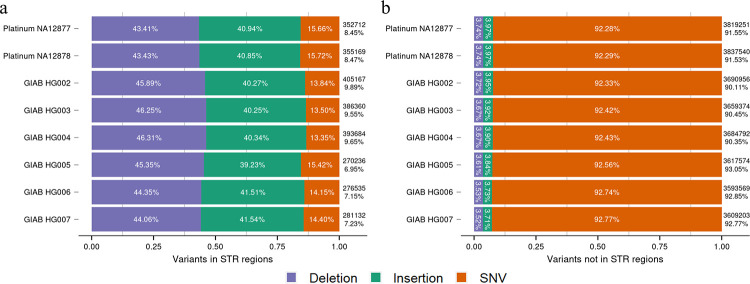
The sequence contexts of genomic variants play important roles in understanding biological significances of variants and potential sequencing related variant calling issues. However, methods for assessing the diverse sequence contexts of genomic variants such as tandem repeats and unambiguous annotations have been limited. Herein, we describe the Variant Sequence Context Annotation Tool (VarSCAT) for annotating the sequence contexts of genomic variants, including breakpoint ambiguities, flanking bases of variants, wildtype/mutated DNA sequences, variant nomenclatures, distances between adjacent variants, tandem repeat regions, and custom annotation with user customizable options. Our analyses demonstrate that VarSCAT is more versatile and customizable than the currently available methods or strategies for annotating variants in short tandem repeat (STR) regions or insertions and deletions (indels) with breakpoint ambiguity. Variant sequence context annotations of high-confidence human variant sets with VarSCAT revealed that more than 75% of all human individual germline and clinically relevant indels have breakpoint ambiguities. Moreover, we illustrate that more than 80% of human individual germline small variants in STR regions are indels and that the sizes of these indels correlated with STR motif sizes. VarSCAT is available from https://github.com/elolab/VarSCAT.
基因组变异的序列背景在理解变异的生物学意义和潜在的测序相关变异调用问题方面起着重要作用。然而,评估基因组变异(如串联重复和明确注释)的各种序列背景的方法一直受到限制。在此,我们描述了用于注释基因组变异序列背景的变体序列上下文注释工具(VarSCAT),包括断点模糊性、变异侧翼碱基、野生型/突变 DNA 序列、变体命名法、相邻变体之间的距离、串联重复区域以及用户可自定义选项的自定义注释。我们的分析表明,VarSCAT 比目前可用的方法或策略更灵活、更具可定制性,可用于注释短串联重复(STR)区域或具有断点模糊性的插入和缺失(indels)中的变体。使用 VarSCAT 对高可信度人类变异集进行变体序列上下文注释表明,超过 75%的所有人类个体种系和临床相关 indels 具有断点模糊性。此外,我们表明,超过 80%的 STR 区域中的人类个体种系小变异是 indels,并且这些 indels 的大小与 STR 基序大小相关。VarSCAT 可从 https://github.com/elolab/VarSCAT 获得。