Department of Cardiology, Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, 310009, Zhejiang, China.
Yuyao People's Hospital of Zhejiang Province, Yuyao, Ningbo, 315400, Zhejiang, China.
BMC Cardiovasc Disord. 2023 Aug 12;23(1):399. doi: 10.1186/s12872-023-03417-2.
Long QT syndrome (LQTS) is one of the primary causes of sudden cardiac death (SCD) in youth. Studies have identified mutations in ion channel genes as key players in the pathogenesis of LQTS. However, the specific etiology in individual families remains unknown.
Three unrelated Chinese pedigrees diagnosed with LQTS or Jervell and Lange-Nielsen syndrome (JLNS) were recruited clinically. Whole exome sequencing (WES) was performed and further validated by multiplex ligation-dependent probe amplification (MLPA) and Sanger sequencing.
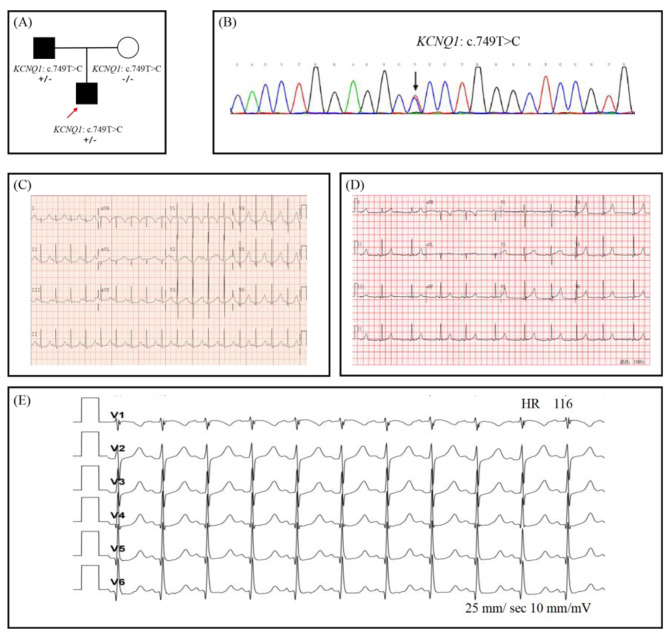
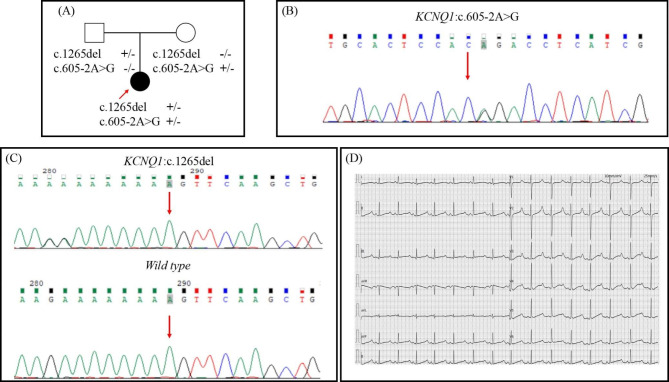
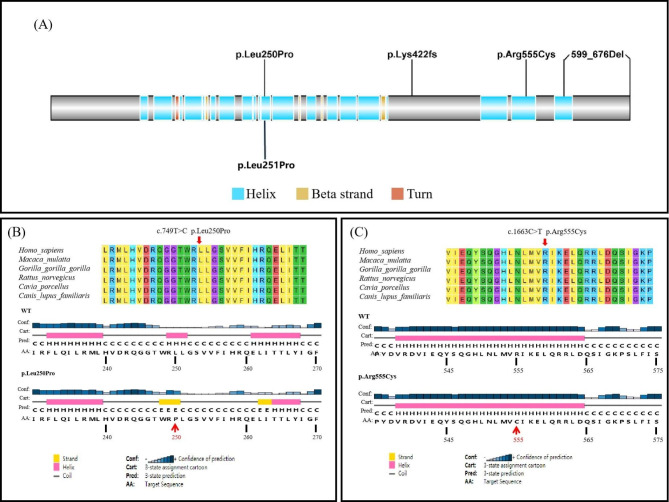
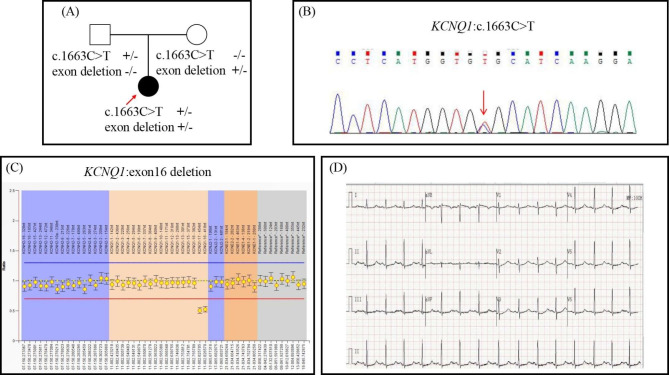
All of the probands in our study experienced syncope episodes and featured typically prolonged QTc-intervals. Two probands also presented with congenital hearing loss and iron-deficiency anemia and thus were diagnosed with JLNS. A total of five different variants in KCNQ1, encoding a subunit of the voltage-gated potassium channel, were identified in 3 probands. The heterozygous variants, KCNQ1 c.749T > C was responsible for LQTS in Case 1, transmitting in an autosomal dominant pattern. Two patterns of compound heterozygous variants were responsible for JLNS, including a large deletion causing loss of the exon 16 and missense variant c.1663 C > T in Case 2, and splicing variant c.605-2 A > G and frame-shift variant c.1265del in Case 3. To our knowledge, the compound heterozygous mutations containing a large deletion and missense variant were first reported in patients with JLNS.
Our study expanded the LQTS genetic spectrum, thus favoring disease screening and diagnosis, personalized treatment, and genetic consultation.
长 QT 综合征(LQTS)是青年人群心源性猝死(SCD)的主要原因之一。研究已经确定离子通道基因突变是 LQTS 发病机制的关键因素。然而,个别家族的具体病因仍不清楚。
临床招募了三个无血缘关系的中国 LQTS 或 Jervell 和 Lange-Nielsen 综合征(JLNS)家系。进行全外显子组测序(WES),并通过多重连接依赖性探针扩增(MLPA)和 Sanger 测序进一步验证。
本研究中的所有先证者均经历晕厥发作,并具有典型的 QT 间期延长。两名先证者还伴有先天性听力损失和缺铁性贫血,因此被诊断为 JLNS。在 3 个先证者中鉴定出编码电压门控钾通道亚基的 KCNQ1 基因中的 5 个不同变异。杂合变异 KCNQ1 c.749T > C 导致 1 号病例的 LQTS,呈常染色体显性遗传模式。2 个复合杂合变异导致 JLNS,包括导致外显子 16 缺失的大片段缺失和 2 号病例中的错义变异 c.1663C > T,以及 3 号病例中的剪接变异 c.605-2A > G 和框移变异 c.1265del。据我们所知,含有大片段缺失和错义变异的复合杂合突变在 JLNS 患者中首次报道。
本研究扩展了 LQTS 的遗传谱,有利于疾病筛查和诊断、个性化治疗和遗传咨询。