Heart Centre, Department of Cardiology, Amsterdam Universitair Medische Centra, Amsterdam, The Netherlands
Heart Centre, Department of Cardiology, Amsterdam Universitair Medische Centra, Amsterdam, The Netherlands.
Heart. 2022 Mar;108(5):332-338. doi: 10.1136/heartjnl-2020-318259. Epub 2021 May 26.
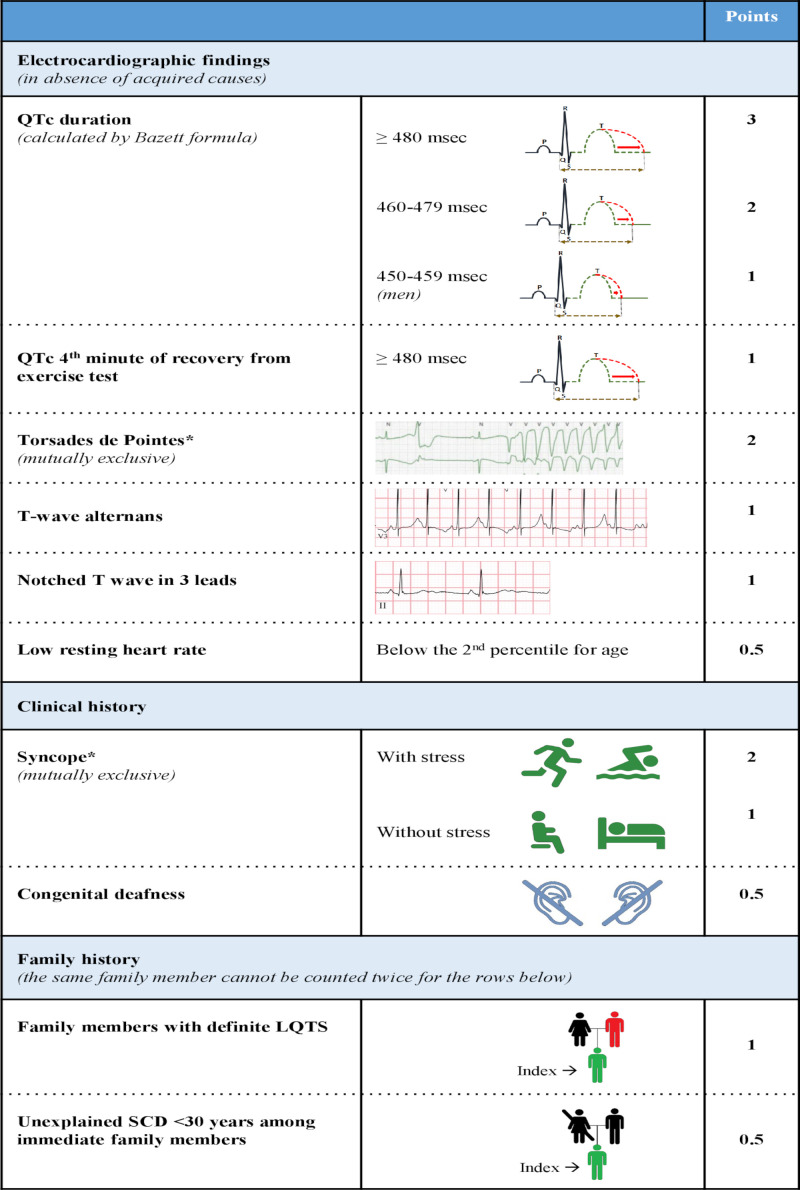
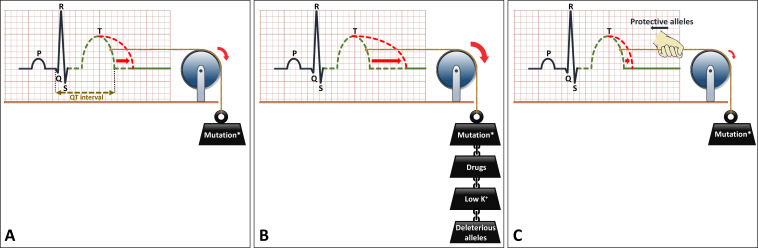
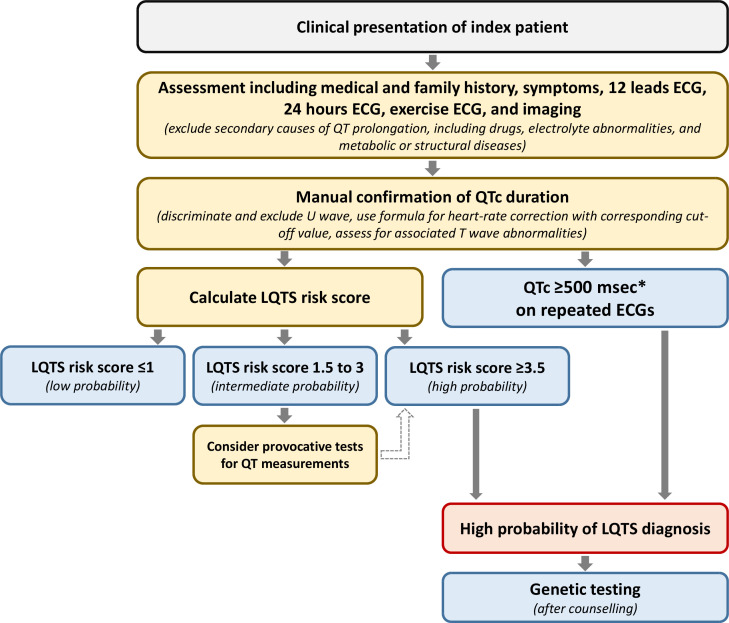
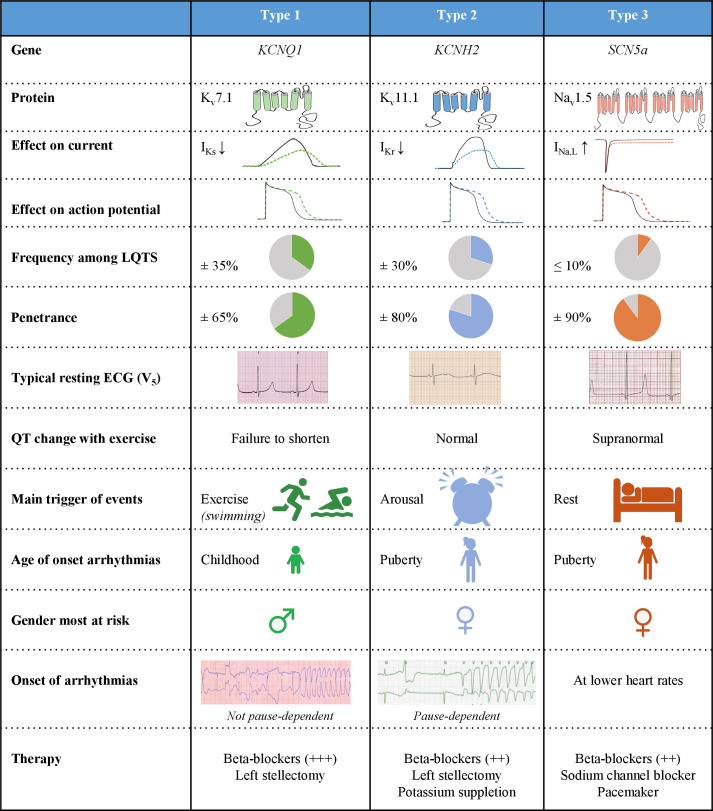
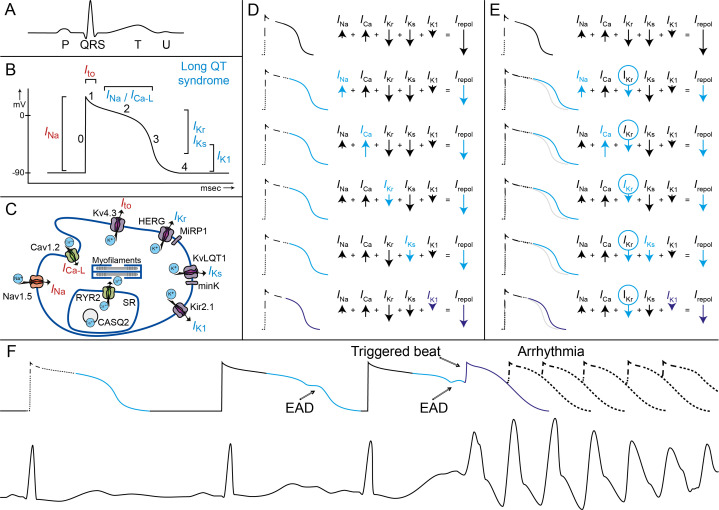
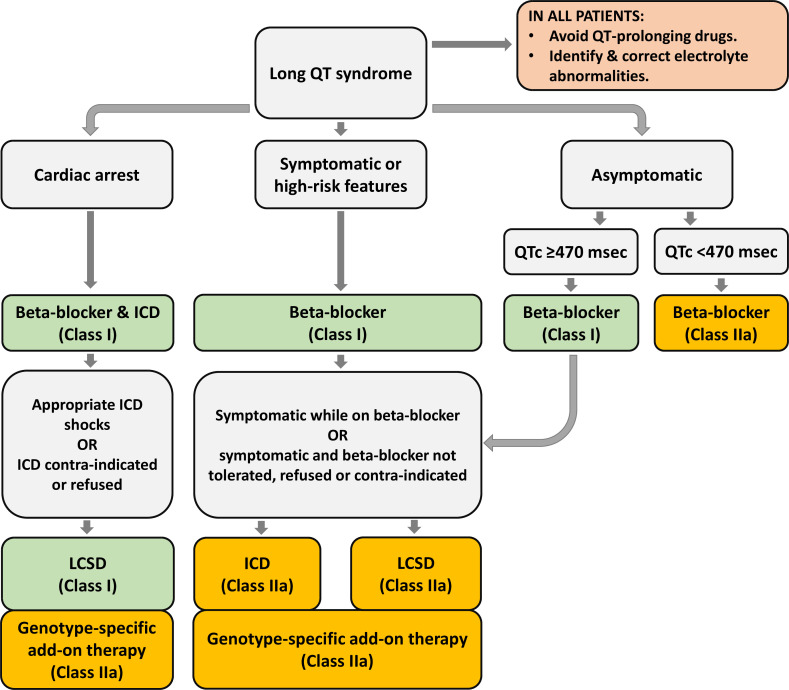
Congenital long QT syndrome (LQTS) is characterised by heart rate corrected QT interval prolongation and life-threatening arrhythmias, leading to syncope and sudden death. Variations in genes encoding for cardiac ion channels, accessory ion channel subunits or proteins modulating the function of the ion channel have been identified as disease-causing mutations in up to 75% of all LQTS cases. Based on the underlying genetic defect, LQTS has been subdivided into different subtypes. Growing insights into the genetic background and pathophysiology of LQTS has led to the identification of genotype-phenotype relationships for the most common genetic subtypes, the recognition of genetic and non-genetic modifiers of phenotype, optimisation of risk stratification algorithms and the discovery of gene-specific therapies in LQTS. Nevertheless, despite these great advancements in the LQTS field, large gaps in knowledge still exist. For example, up to 25% of LQTS cases still remain genotype elusive, which hampers proper identification of family members at risk, and it is still largely unknown what determines the large variability in disease severity, where even within one family an identical mutation causes malignant arrhythmias in some carriers, while in other carriers, the disease is clinically silent. In this review, we summarise the current evidence available on the diagnosis, clinical management and therapeutic strategies in LQTS. We also discuss new scientific developments and areas of research, which are expected to increase our understanding of the complex genetic architecture in genotype-negative patients, lead to improved risk stratification in asymptomatic mutation carriers and more targeted (gene-specific and even mutation-specific) therapies.
先天性长 QT 综合征(LQTS)的特征是心率校正 QT 间期延长和危及生命的心律失常,导致晕厥和猝死。编码心脏离子通道、辅助离子通道亚基或调节离子通道功能的蛋白质的基因突变已被确定为高达 75%的所有 LQTS 病例的致病突变。根据潜在的遗传缺陷,LQTS 已分为不同的亚型。对 LQTS 的遗传背景和病理生理学的深入了解导致了最常见遗传亚型的基因型-表型关系的确定、表型的遗传和非遗传修饰因子的识别、风险分层算法的优化以及 LQTS 中基因特异性治疗的发现。然而,尽管在 LQTS 领域取得了这些重大进展,但仍存在许多知识空白。例如,高达 25%的 LQTS 病例仍然无法确定基因型,这阻碍了对有风险的家族成员的正确识别,而且是什么决定了疾病严重程度的巨大变异性仍然很大程度上未知,即使在一个家族中,相同的突变也会导致一些携带者发生恶性心律失常,而在其他携带者中,疾病则没有临床症状。在这篇综述中,我们总结了目前关于 LQTS 的诊断、临床管理和治疗策略的证据。我们还讨论了新的科学发展和研究领域,预计这将增加我们对基因型阴性患者复杂遗传结构的理解,导致无症状突变携带者的风险分层得到改善,并提供更有针对性的(基因特异性甚至突变特异性)治疗。