Department of Integrative Biology, University of Texas at Austin, Austin, TX, USA.
The Department of Molecular Biosciences, Center for Systems and Synthetic Biology, The University of Texas at Austin, Austin, TX, USA.
Sci Rep. 2023 Aug 16;13(1):13280. doi: 10.1038/s41598-023-40247-w.
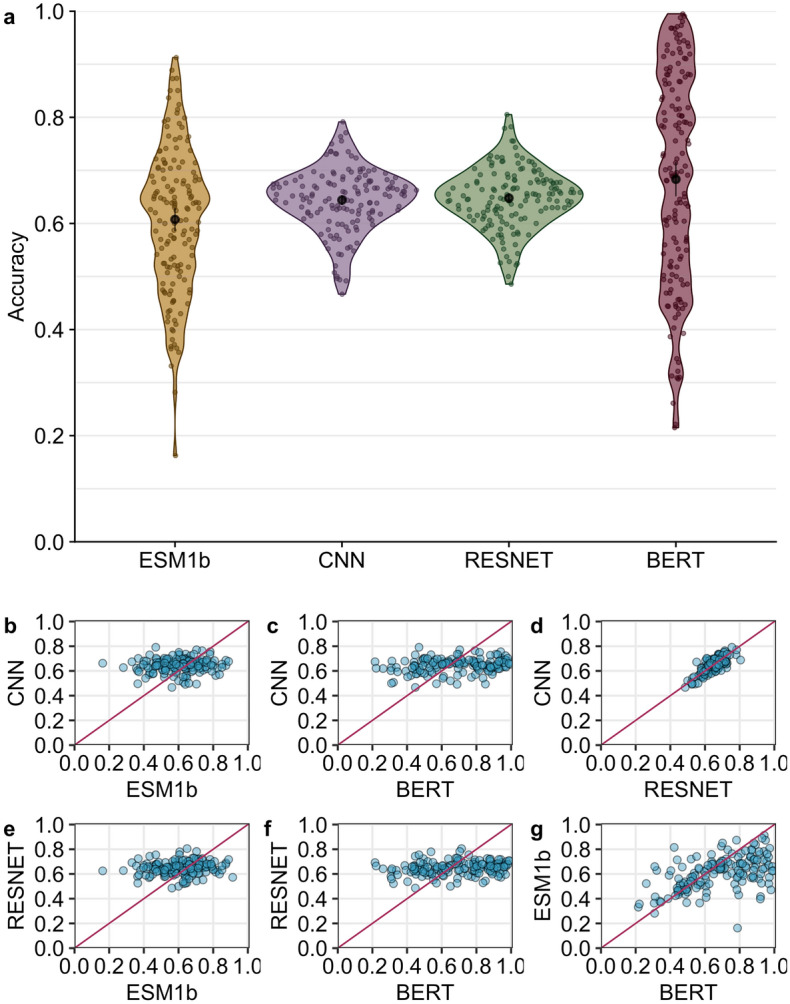
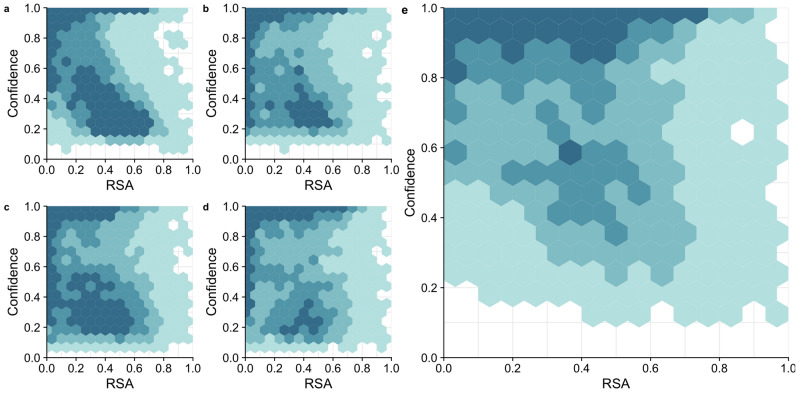
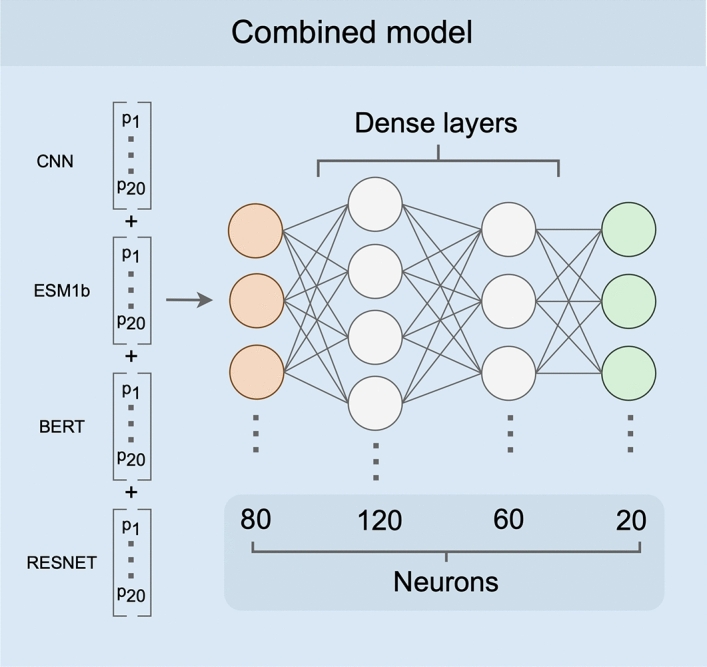
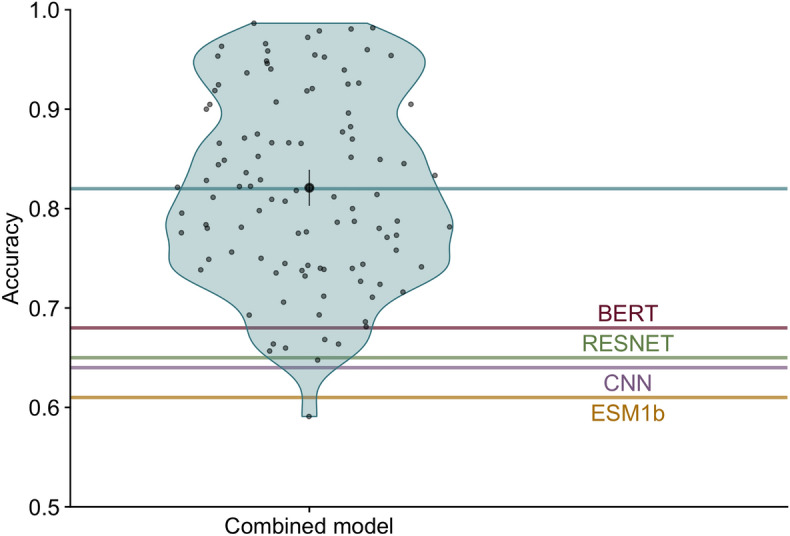
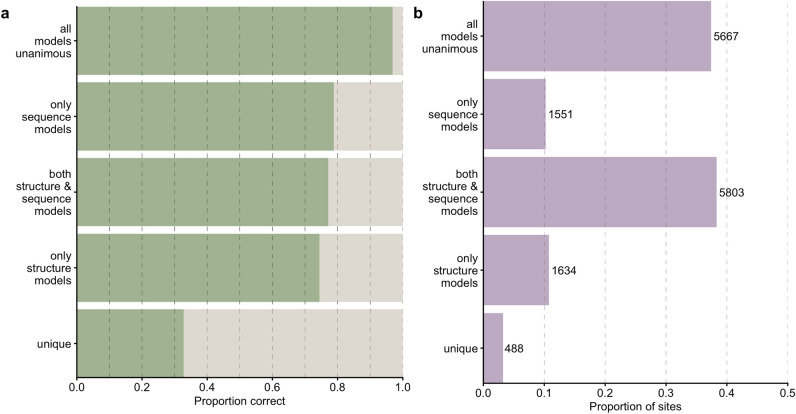
Deep learning models are seeing increased use as methods to predict mutational effects or allowed mutations in proteins. The models commonly used for these purposes include large language models (LLMs) and 3D Convolutional Neural Networks (CNNs). These two model types have very different architectures and are commonly trained on different representations of proteins. LLMs make use of the transformer architecture and are trained purely on protein sequences whereas 3D CNNs are trained on voxelized representations of local protein structure. While comparable overall prediction accuracies have been reported for both types of models, it is not known to what extent these models make comparable specific predictions and/or generalize protein biochemistry in similar ways. Here, we perform a systematic comparison of two LLMs and two structure-based models (CNNs) and show that the different model types have distinct strengths and weaknesses. The overall prediction accuracies are largely uncorrelated between the sequence- and structure-based models. Overall, the two structure-based models are better at predicting buried aliphatic and hydrophobic residues whereas the two LLMs are better at predicting solvent-exposed polar and charged amino acids. Finally, we find that a combined model that takes the individual model predictions as input can leverage these individual model strengths and results in significantly improved overall prediction accuracy.
深度学习模型作为预测蛋白质突变效应或允许突变的方法越来越受到关注。这些目的常用的模型包括大型语言模型(LLM)和 3D 卷积神经网络(CNN)。这两种模型类型具有非常不同的架构,通常在蛋白质的不同表示形式上进行训练。LLM 利用转换器架构,仅在蛋白质序列上进行训练,而 3D CNN 则在局部蛋白质结构的体素化表示上进行训练。虽然两种类型的模型都报告了相当的总体预测准确性,但尚不清楚这些模型在多大程度上可以进行可比的具体预测,以及以类似的方式推广蛋白质生物化学。在这里,我们对两个 LLM 和两个基于结构的模型(CNN)进行了系统比较,并表明不同的模型类型具有不同的优缺点。基于序列和基于结构的模型之间的总体预测准确性相关性不大。总体而言,两种基于结构的模型更擅长预测埋藏的脂肪族和亲脂性残基,而两种 LLM 更擅长预测溶剂暴露的极性和带电氨基酸。最后,我们发现,将个别模型预测作为输入的组合模型可以利用这些个别模型的优势,并显著提高整体预测准确性。