Advanced Biomedical Computational Science, Frederick National Laboratory for Cancer Research, Frederick, MD, 21702, USA.
Division of Preclinical Innovation, Therapeutic Development Branch, Therapeutics for Rare and Neglected Diseases (TRND) Program, National Center for Advancing Translational Sciences, National Institutes of Health, Bethesda, MD, 20892, USA.
BMC Genomics. 2023 Aug 16;24(1):460. doi: 10.1186/s12864-023-09561-5.
Approximately 4-8% of the world suffers from a rare disease. Rare diseases are often difficult to diagnose, and many do not have approved therapies. Genetic sequencing has the potential to shorten the current diagnostic process, increase mechanistic understanding, and facilitate research on therapeutic approaches but is limited by the difficulty of novel variant pathogenicity interpretation and the communication of known causative variants. It is unknown how many published rare disease variants are currently accessible in the public domain.
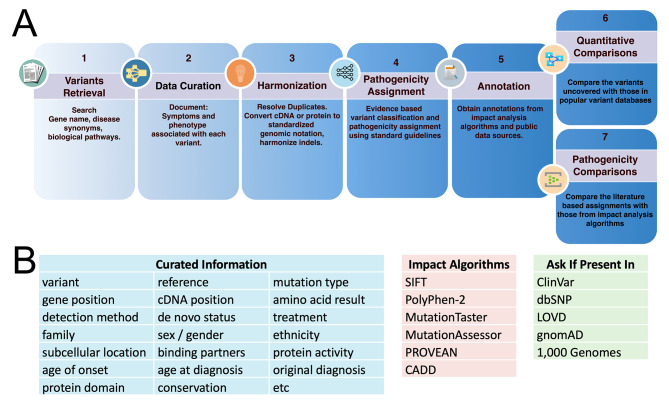
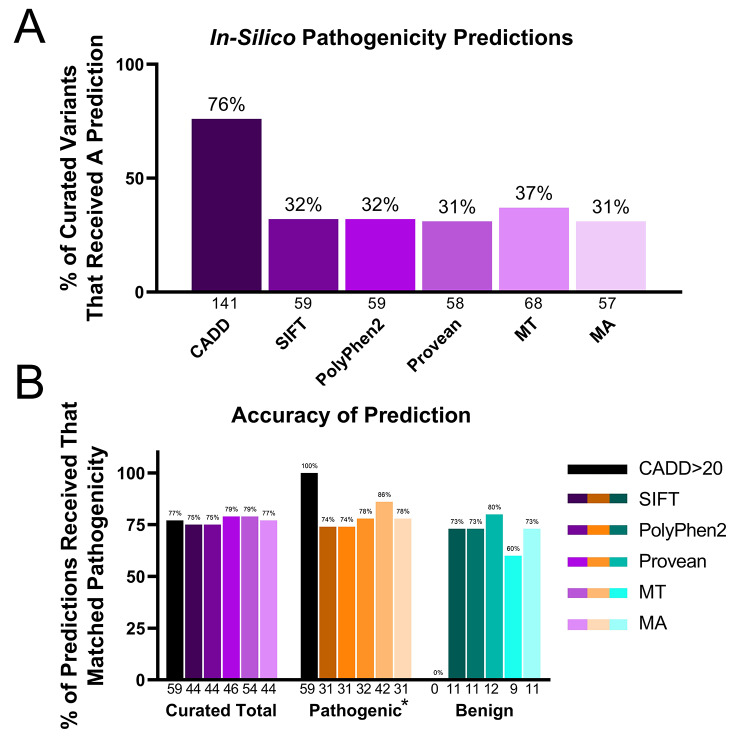
This study investigated the translation of knowledge of variants reported in published manuscripts to publicly accessible variant databases. Variants, symptoms, biochemical assay results, and protein function from literature on the SLC6A8 gene associated with X-linked Creatine Transporter Deficiency (CTD) were curated and reported as a highly annotated dataset of variants with clinical context and functional details. Variants were harmonized, their availability in existing variant databases was analyzed and pathogenicity assignments were compared with impact algorithm predictions. 24% of the pathogenic variants found in PubMed articles were not captured in any database used in this analysis while only 65% of the published variants received an accurate pathogenicity prediction from at least one impact prediction algorithm.
Despite being published in the literature, pathogenicity data on patient variants may remain inaccessible for genetic diagnosis, therapeutic target identification, mechanistic understanding, or hypothesis generation. Clinical and functional details presented in the literature are important to make pathogenicity assessments. Impact predictions remain imperfect but are improving, especially for single nucleotide exonic variants, however such predictions are less accurate or unavailable for intronic and multi-nucleotide variants. Developing text mining workflows that use natural language processing for identifying diseases, genes and variants, along with impact prediction algorithms and integrating with details on clinical phenotypes and functional assessments might be a promising approach to scale literature mining of variants and assigning correct pathogenicity. The curated variants list created by this effort includes context details to improve any such efforts on variant curation for rare diseases.
大约有 4-8%的世界人口患有罕见病。罕见病通常难以诊断,并且许多疾病没有批准的治疗方法。基因测序有可能缩短当前的诊断过程,增加对发病机制的理解,并促进治疗方法的研究,但受到新型变异致病性解释的困难以及已知致病变异的传播的限制。目前尚不清楚有多少已发表的罕见病变异可在公共领域获得。
本研究调查了将发表的手稿中报告的变异知识转化为可公开访问的变异数据库的情况。对与 X 连锁肌酸转运蛋白缺乏症(CTD)相关的 SLC6A8 基因的文献中的变异、症状、生化测定结果和蛋白质功能进行了整理,并以具有临床背景和功能细节的高度注释的变异数据集进行了报道。对变异进行了协调,分析了它们在现有变异数据库中的可用性,并将致病性预测与影响算法预测进行了比较。在 PubMed 文章中发现的致病性变异中有 24%未在本分析中使用的任何数据库中捕获,而只有 65%的已发表变异至少从一种影响预测算法中获得了准确的致病性预测。
尽管已在文献中发表,但患者变异的致病性数据可能仍无法用于遗传诊断、治疗靶点识别、发病机制理解或假说生成。文献中提供的临床和功能细节对于进行致病性评估很重要。影响预测仍然不完美,但正在改进,尤其是对于单核苷酸外显子变异,但对于内含子和多核苷酸变异,此类预测的准确性较低或不可用。开发使用自然语言处理识别疾病、基因和变异的文本挖掘工作流程,以及影响预测算法,并结合临床表型和功能评估的详细信息,可能是一种有前途的方法,可以大规模进行文献挖掘变异并正确分配致病性。本研究创建的经过整理的变异列表包含上下文详细信息,可用于改进罕见病变异的整理工作。