Research and Development Center, 3billion, 14th floor, 416 Teheran-ro, Gangnam-gu, Seoul, 06193, Republic of Korea.
Hum Genomics. 2024 Mar 21;18(1):28. doi: 10.1186/s40246-024-00595-8.
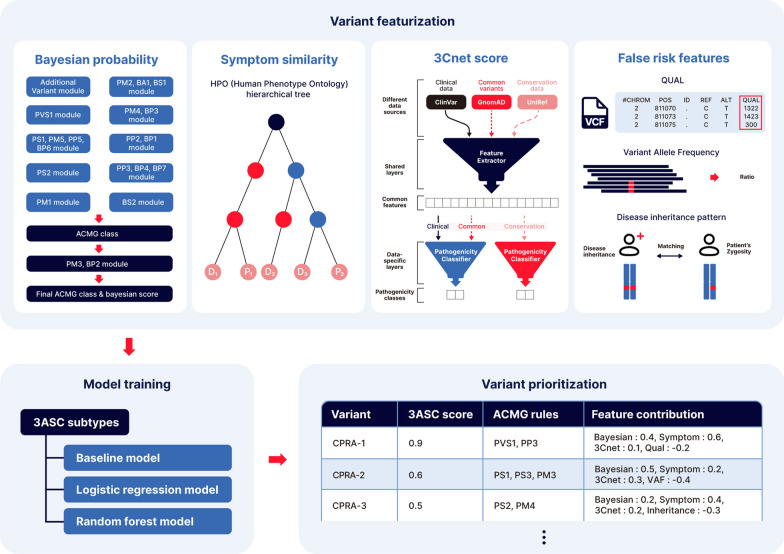
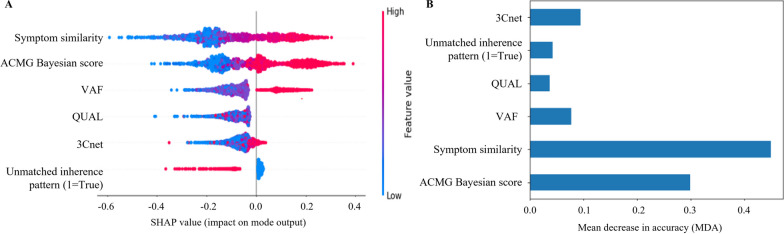
In the process of finding the causative variant of rare diseases, accurate assessment and prioritization of genetic variants is essential. Previous variant prioritization tools mainly depend on the in-silico prediction of the pathogenicity of variants, which results in low sensitivity and difficulty in interpreting the prioritization result. In this study, we propose an explainable algorithm for variant prioritization, named 3ASC, with higher sensitivity and ability to annotate evidence used for prioritization. 3ASC annotates each variant with the 28 criteria defined by the ACMG/AMP genome interpretation guidelines and features related to the clinical interpretation of the variants. The system can explain the result based on annotated evidence and feature contributions.
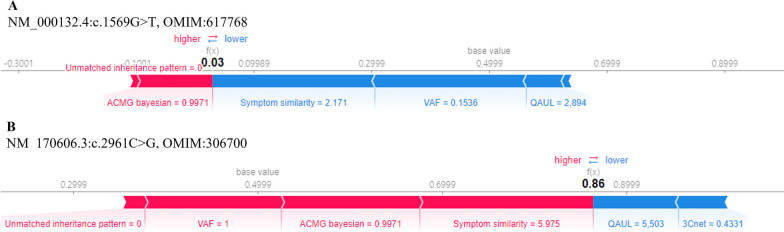
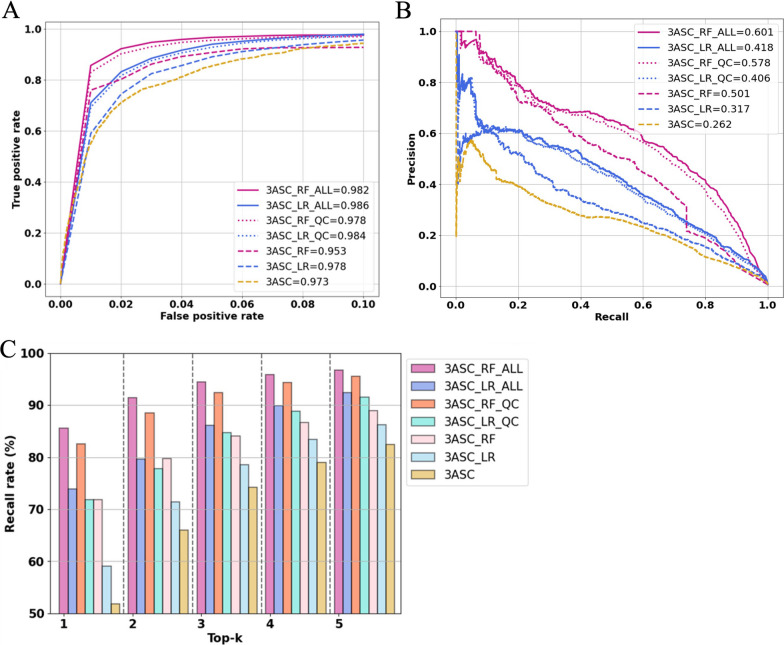
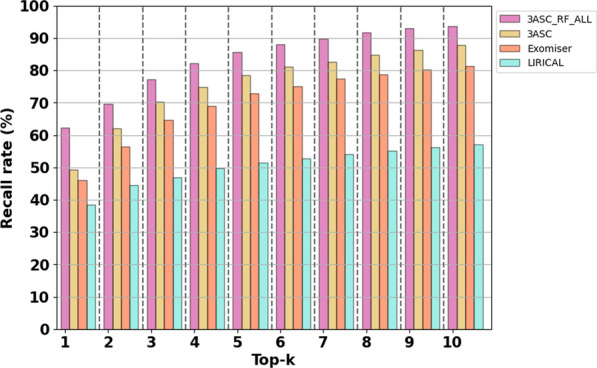
We trained various machine learning algorithms using in-house patient data. The performance of variant ranking was assessed using the recall rate of identifying causative variants in the top-ranked variants. The best practice model was a random forest classifier that showed top 1 recall of 85.6% and top 3 recall of 94.4%. The 3ASC annotates the ACMG/AMP criteria for each genetic variant of a patient so that clinical geneticists can interpret the result as in the CAGI6 SickKids challenge. In the challenge, 3ASC identified causal genes for 10 out of 14 patient cases, with evidence of decreased gene expression for 6 cases. Among them, two genes (HDAC8 and CASK) had decreased gene expression profiles confirmed by transcriptome data.
3ASC can prioritize genetic variants with higher sensitivity compared to previous methods by integrating various features related to clinical interpretation, including features related to false positive risk such as quality control and disease inheritance pattern. The system allows interpretation of each variant based on the ACMG/AMP criteria and feature contribution assessed using explainable AI techniques.
在寻找罕见病致病变体的过程中,准确评估和优先考虑遗传变体至关重要。以前的变体优先级工具主要依赖于变体致病性的计算机预测,这导致敏感性低,并且难以解释优先级结果。在这项研究中,我们提出了一种可解释的变体优先级算法,名为 3ASC,它具有更高的敏感性和注释优先级使用证据的能力。3ASC 为每个变体标注了 ACMG/AMP 基因组解释指南定义的 28 个标准以及与变体临床解释相关的特征。系统可以根据标注的证据和特征贡献来解释结果。
我们使用内部患者数据训练了各种机器学习算法。变体排名的性能通过在排名靠前的变体中识别致病变体的召回率来评估。最佳实践模型是随机森林分类器,其前 1 名召回率为 85.6%,前 3 名召回率为 94.4%。3ASC 为患者的每个遗传变体标注了 ACMG/AMP 标准,以便临床遗传学家可以像在 CAGI6 SickKids 挑战赛中那样解释结果。在挑战赛中,3ASC 确定了 14 个患者案例中的 10 个因果基因,其中 6 个有基因表达降低的证据。其中,两个基因(HDAC8 和 CASK)的基因表达谱降低情况得到了转录组数据的证实。
与以前的方法相比,3ASC 通过整合与临床解释相关的各种特征,包括与假阳性风险相关的特征(如质量控制和疾病遗传模式),可以以更高的敏感性对遗传变体进行优先级排序。该系统允许根据 ACMG/AMP 标准和使用可解释 AI 技术评估的特征贡献来解释每个变体。