Department of Clinical Medicine, Aarhus University, Palle Juul-Jensens Boulevard 82, 8200, Aarhus N, Denmark.
Department of Molecular Medicine (MOMA), Aarhus University Hospital, Palle Juul-Jensens Boulevard 99, 8200, Aarhus N, Denmark.
Genome Med. 2023 Aug 17;15(1):63. doi: 10.1186/s13073-023-01217-z.
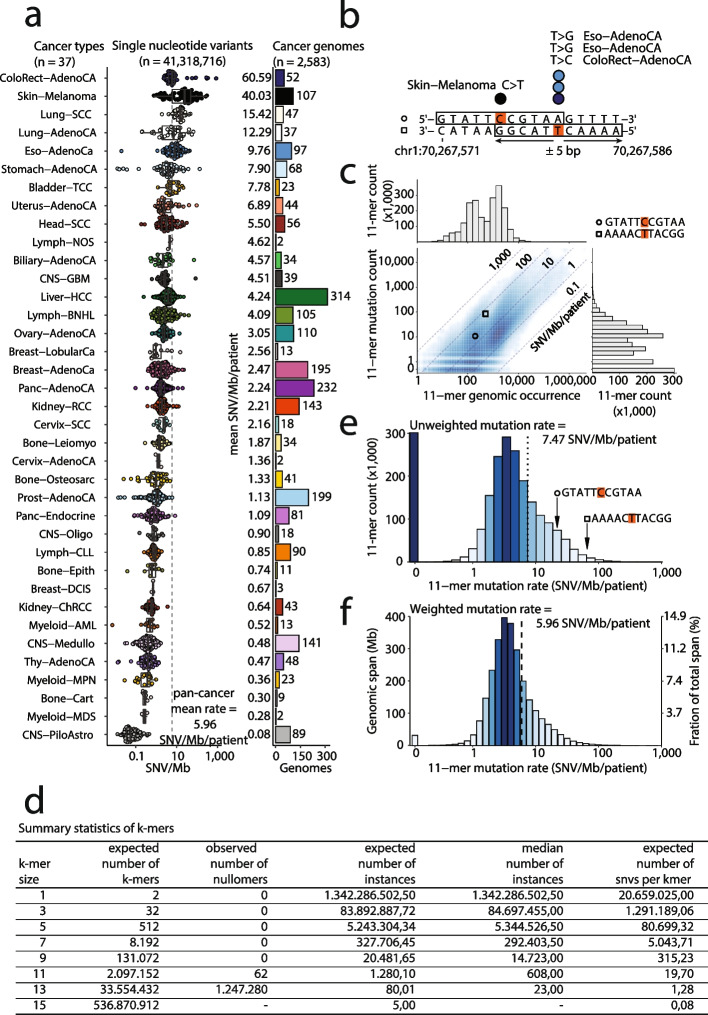
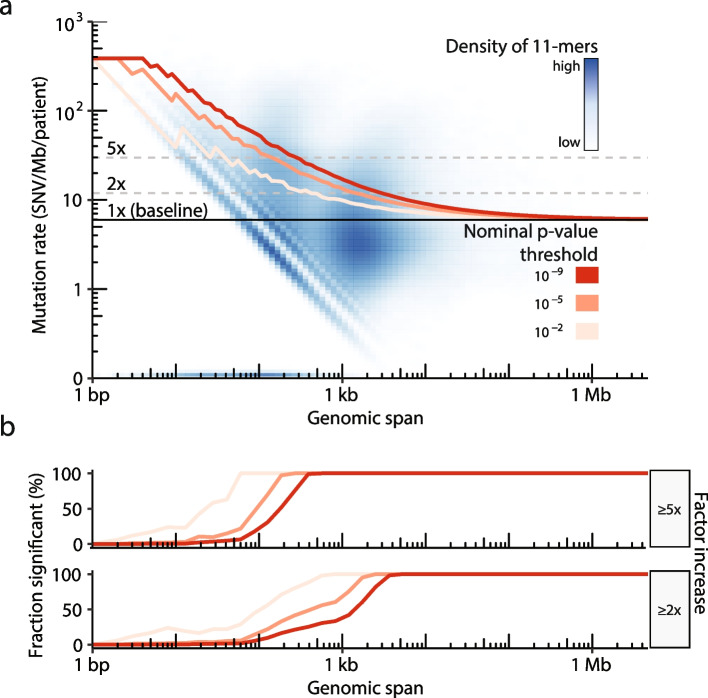
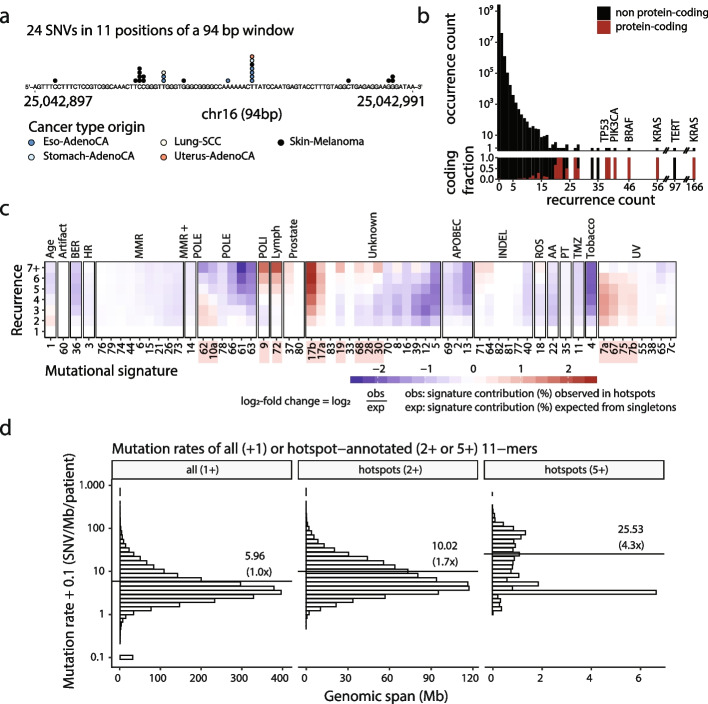
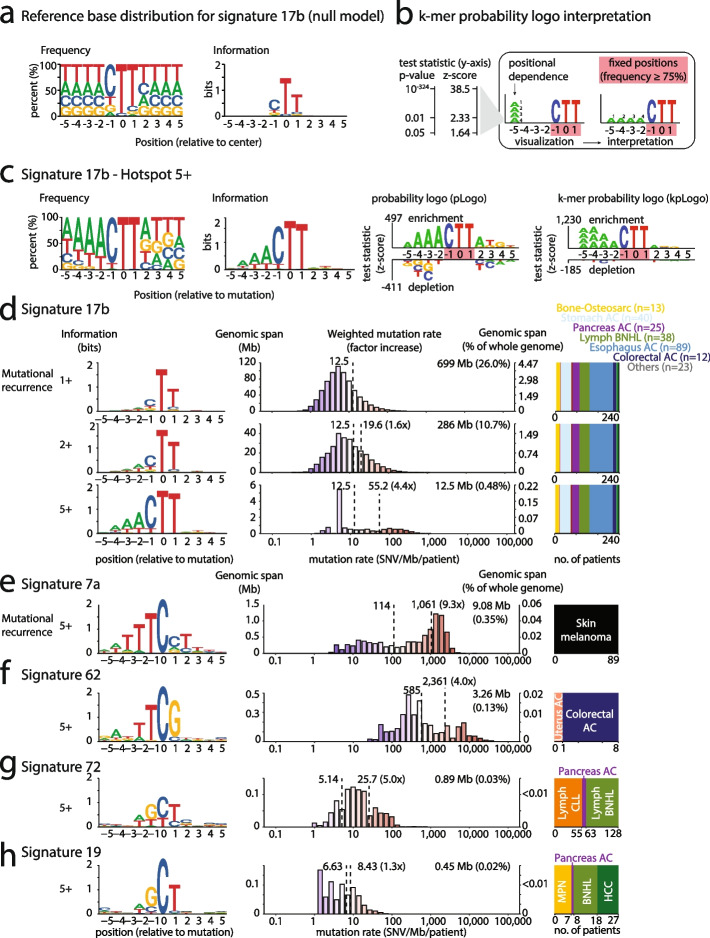
Cancer mutations accumulate through replication errors and DNA damage coupled with incomplete repair. Individual mutational processes often show nucleotide sequence and functional region preferences. As a result, some sequence contexts mutate at much higher rates than others, with additional variation found between functional regions. Mutational hotspots, with recurrent mutations across cancer samples, represent genomic positions with elevated mutation rates, often caused by highly localized mutational processes.
We count the 11-mer genomic sequences across the genome, and using the PCAWG set of 2583 pan-cancer whole genomes, we associate 11-mers with mutational signatures, hotspots of single nucleotide variants, and specific genomic regions. We evaluate the mutation rates of individual and combined sets of 11-mers and derive mutational sequence motifs.
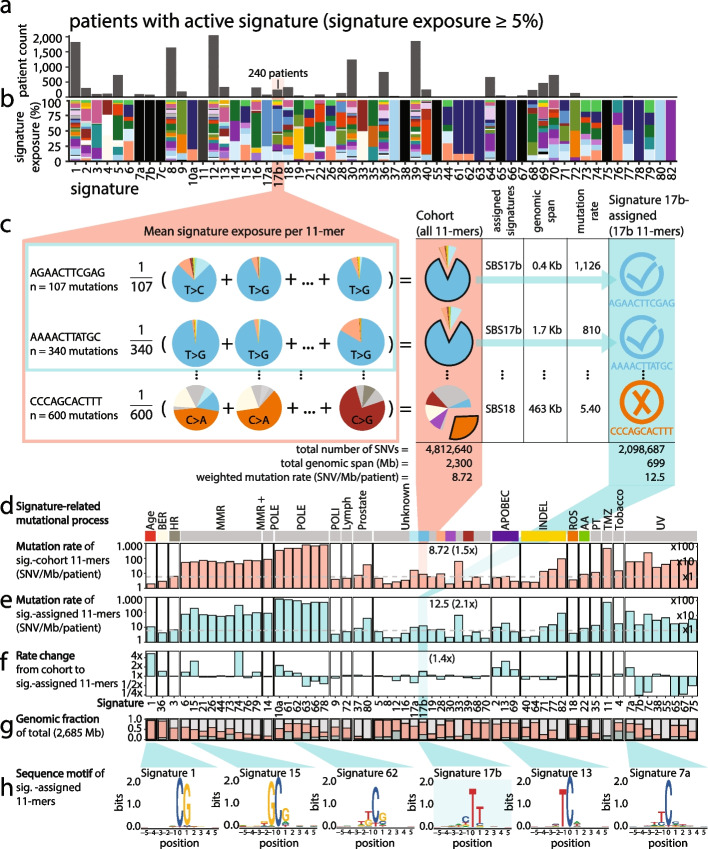
We show that hotspots generally identify highly mutable sequence contexts. Using these, we show that some mutational signatures are enriched in hotspot sequence contexts, corresponding to well-defined sequence preferences for the underlying localized mutational processes. This includes signature 17b (of unknown etiology) and signatures 62 (POLE deficiency), 7a (UV), and 72 (linked to lymphomas). In some cases, the mutation rate and sequence preference increase further when focusing on certain genomic regions, such as signature 62 in transcribed regions, where the mutation rate is increased up to 9-folds over cancer type and mutational signature average.
We summarize our findings in a catalog of localized mutational processes, their sequence preferences, and their estimated mutation rates.
癌症突变是通过复制错误和 DNA 损伤以及不完全修复而积累的。个体突变过程通常表现出核苷酸序列和功能区域的偏好。因此,一些序列背景的突变率比其他序列背景高得多,在功能区域之间还存在额外的变化。突变热点是指在癌症样本中经常出现的反复突变的基因组位置,代表了突变率升高的基因组位置,通常是由高度局部化的突变过程引起的。
我们在整个基因组上计算 11-mer 基因组序列,并使用 PCAWG 中的 2583 个泛癌症全基因组,将 11-mer 与突变特征、单核苷酸变异热点和特定基因组区域相关联。我们评估了单个和组合的 11- mer 的突变率,并推导出突变序列基序。
我们表明热点通常可以识别高度易变的序列背景。利用这些热点,我们表明一些突变特征在热点序列背景中富集,对应于潜在局部突变过程的明确序列偏好。这包括特征 17b(病因不明)和特征 62(POLE 缺陷)、7a(UV)和 72(与淋巴瘤相关)。在某些情况下,当关注某些基因组区域时,突变率和序列偏好会进一步增加,例如特征 62 在转录区域,突变率比癌症类型和突变特征平均高出 9 倍。
我们将发现总结在本地化突变过程、其序列偏好及其估计的突变率目录中。