Juul Randi Istrup, Nielsen Morten Muhlig, Juul Malene, Feuerbach Lars, Pedersen Jakob Skou
Department of Molecular Medicine, Aarhus University Hospital, Aarhus, Denmark.
Division of Applied Bioinformatics, German Cancer Research Center, Heidelberg, Germany.
NPJ Genom Med. 2021 May 13;6(1):33. doi: 10.1038/s41525-021-00197-6.
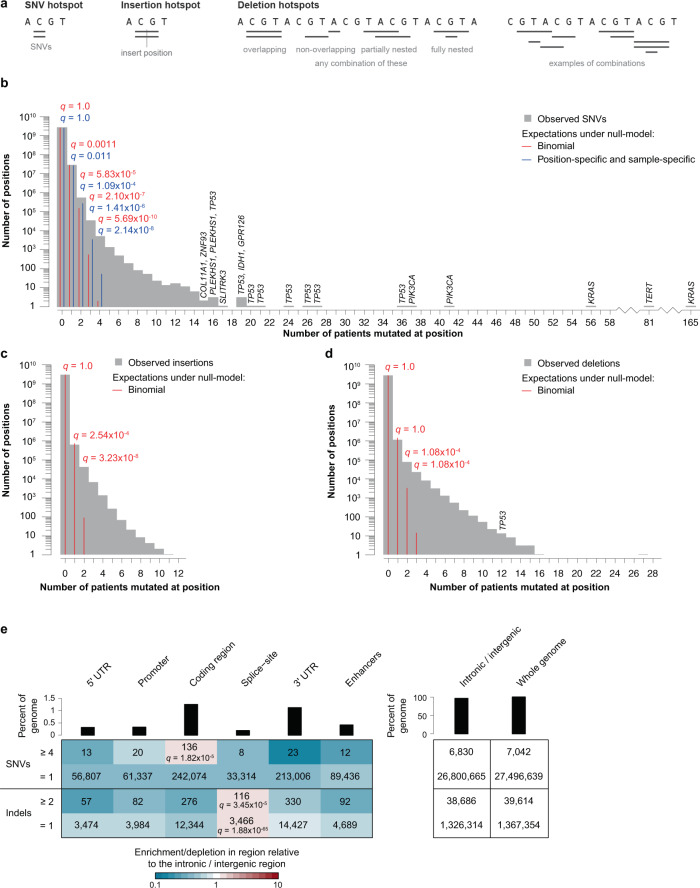
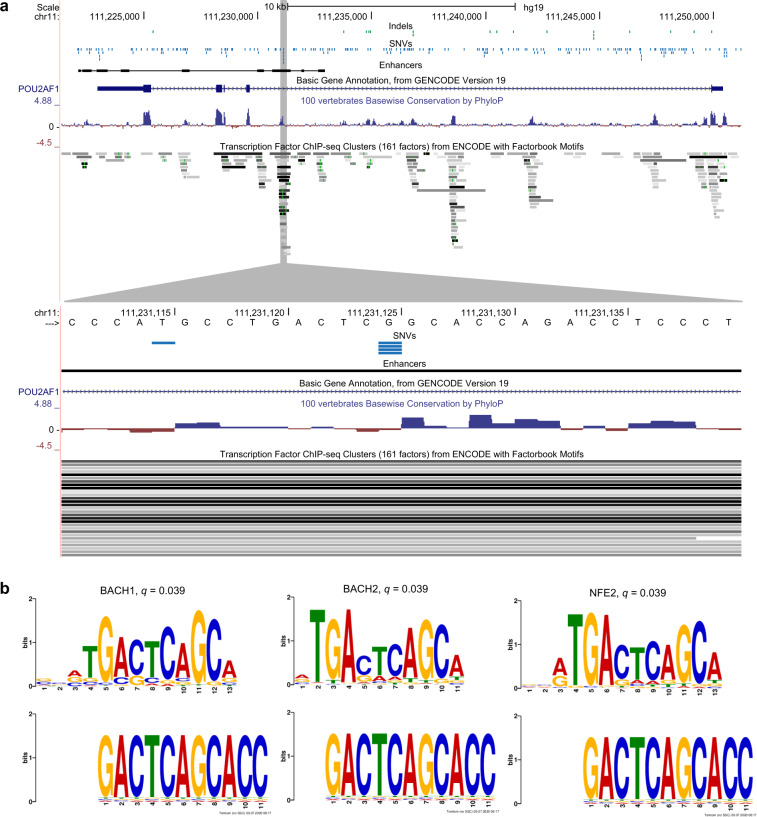
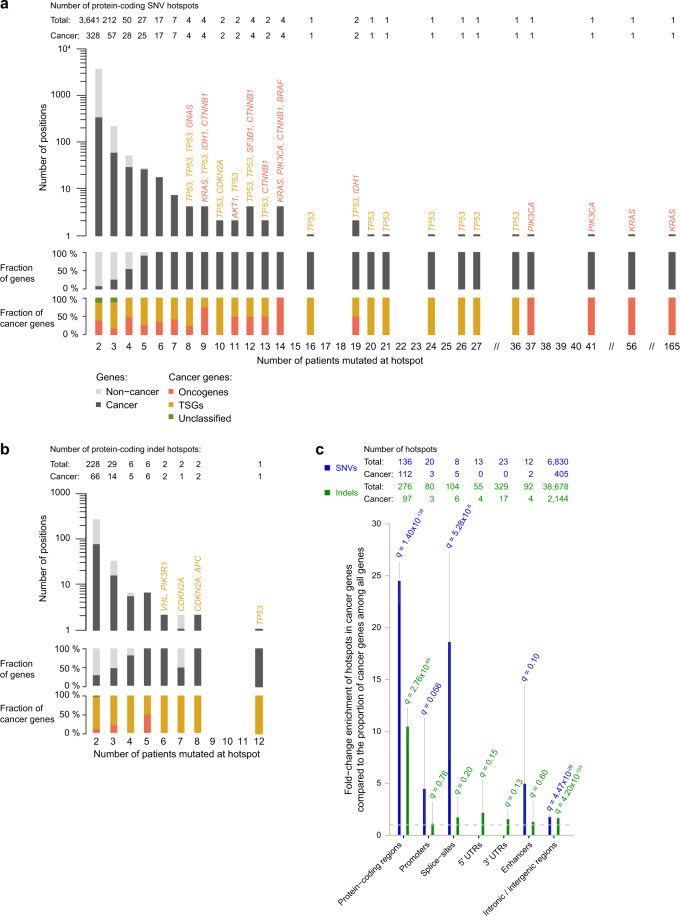
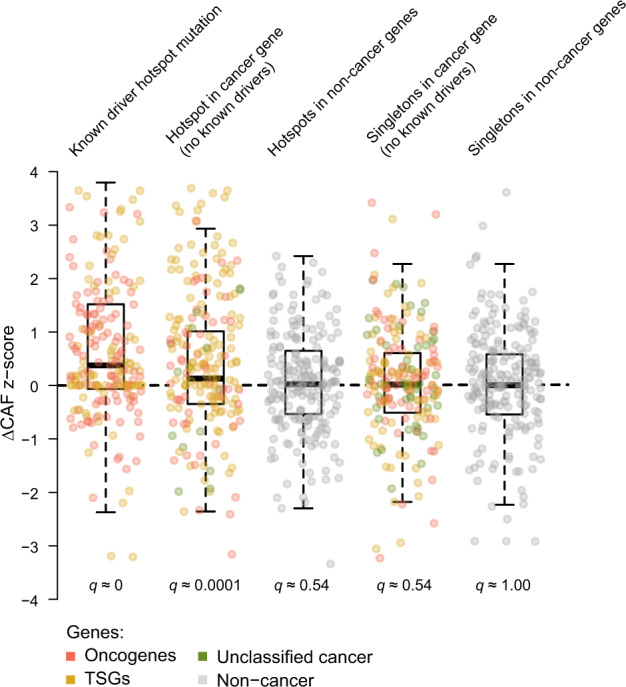
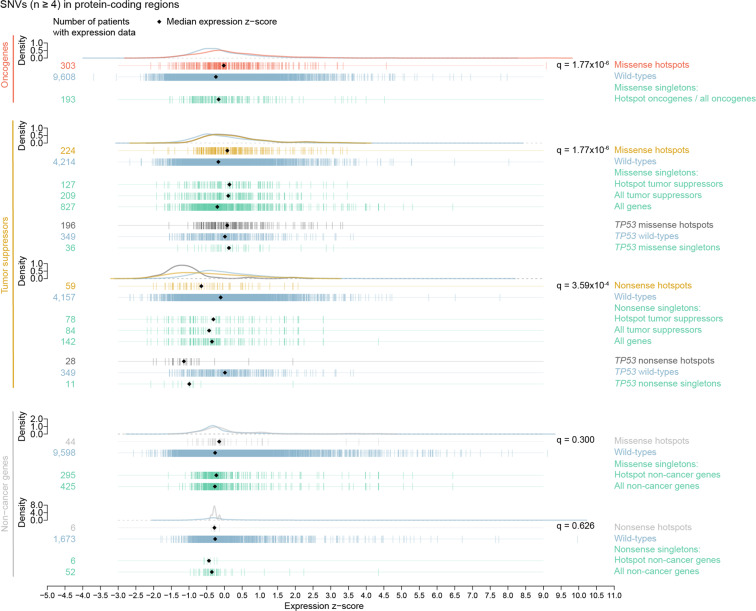
Large sets of whole cancer genomes make it possible to study mutation hotspots genome-wide. Here we detect, categorize, and characterize site-specific hotspots using 2279 whole cancer genomes from the Pan-Cancer Analysis of Whole Genomes project and provide a resource of annotated hotspots genome-wide. We investigate the excess of hotspots in both protein-coding and gene regulatory regions and develop measures of positive selection and functional impact for individual hotspots. Using cancer allele fractions, expression aberrations, mutational signatures, and a variety of genomic features, such as potential gain or loss of transcription factor binding sites, we annotate and prioritize all highly mutated hotspots. Genome-wide we find more high-frequency SNV and indel hotspots than expected given mutational background models. Protein-coding regions are generally enriched for SNV hotspots compared to other regions. Gene regulatory hotspots show enrichment of potential same-patient second-hit missense mutations, consistent with enrichment of hotspot driver mutations compared to singletons. For protein-coding regions, splice-sites, promoters, and enhancers, we see an excess of hotspots associated with cancer genes. Interestingly, missense hotspot mutations in tumor suppressors are associated with elevated expression, suggesting localized amino-acid changes with functional impact. For individual non-coding hotspots, only a small number show clear signs of positive selection, including known sites in the TERT promoter and the 5' UTR of TP53. Most of the new candidates have few mutations and limited driver evidence. However, a hotspot in an enhancer of the oncogene POU2AF1, which may create a transcription factor binding site, presents multiple lines of driver-consistent evidence.
大量的全癌基因组使得在全基因组范围内研究突变热点成为可能。在这里,我们使用来自全基因组泛癌分析项目的2279个全癌基因组来检测、分类和表征位点特异性热点,并提供全基因组注释热点的资源。我们研究了蛋白质编码区和基因调控区中热点的过量情况,并开发了针对单个热点的正选择和功能影响的测量方法。利用癌症等位基因分数、表达异常、突变特征以及各种基因组特征,如转录因子结合位点的潜在增减,我们对所有高度突变的热点进行注释并排序。在全基因组范围内,我们发现高频单核苷酸变异(SNV)和插入缺失热点比基于突变背景模型预期的更多。与其他区域相比,蛋白质编码区通常富含SNV热点。基因调控热点显示出潜在的同一患者二次打击错义突变的富集,这与热点驱动突变相对于单例突变的富集一致。对于蛋白质编码区、剪接位点、启动子和增强子,我们发现与癌症基因相关的热点过量。有趣的是,肿瘤抑制基因中的错义热点突变与表达升高相关,表明局部氨基酸变化具有功能影响。对于单个非编码热点,只有少数显示出明显的正选择迹象,包括端粒酶逆转录酶(TERT)启动子和TP53的5'非翻译区(UTR)中的已知位点。大多数新候选热点的突变很少且驱动证据有限。然而,致癌基因POU2AF1增强子中的一个热点可能会产生一个转录因子结合位点,它呈现出多条与驱动一致的证据。