Department of Pediatrics, "G D'Annunzioof Pediatrics, " University of Chieti-Pescara, Foggia, Italy.
Unit of Endocrinology, Fondazione Istituto di Ricerca e Cura a Carattere Scientifico (IRCCS) Casa Sollievo della Sofferenza, Foggia, Italy.
Front Endocrinol (Lausanne). 2023 Aug 1;14:1205977. doi: 10.3389/fendo.2023.1205977. eCollection 2023.
Hypophosphatasia (HPP) is a rare genetic disease caused by inactivating variants of the ALPL gene. Few data are available on the clinical presentation in Italy and/or on Italian HPP surveys.
There were 30 suspected HPP patients recruited from different Italian tertiary cares. Biological samples and related clinical, biochemical, and anamnestic data were collected and the ALPL gene sequenced. Search for large genomic deletions at the ALPL locus (1p36) was done. Phylogenetic conservation and modeling were applied to infer the effect of the variants on the protein structure.
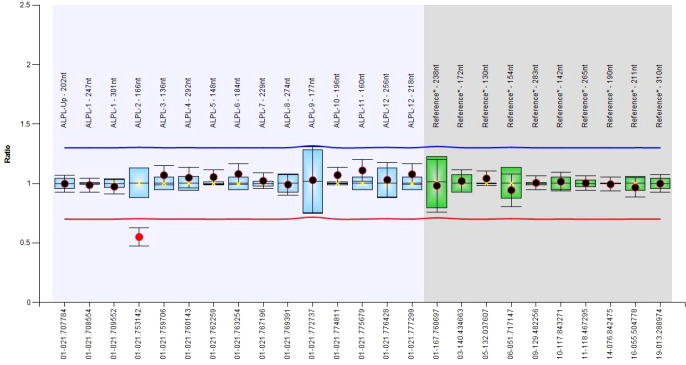
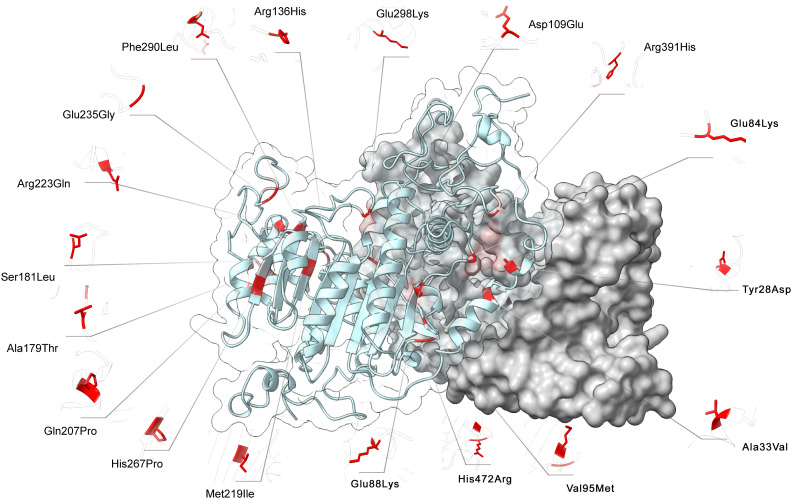
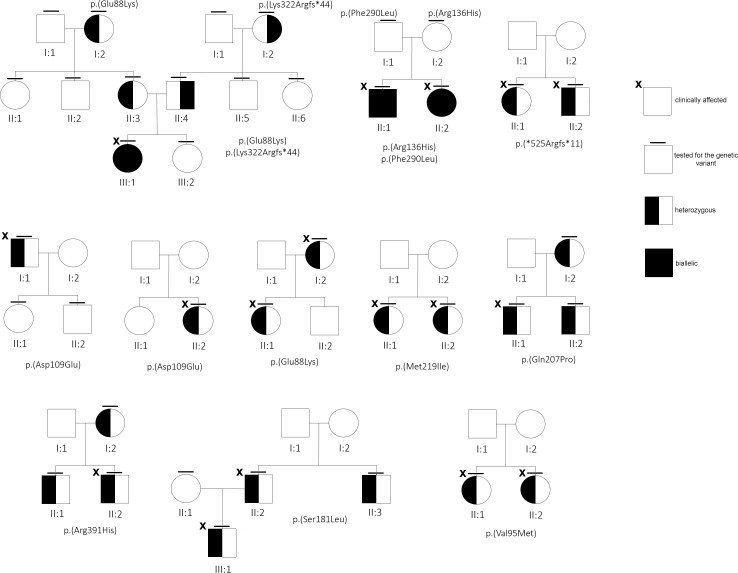
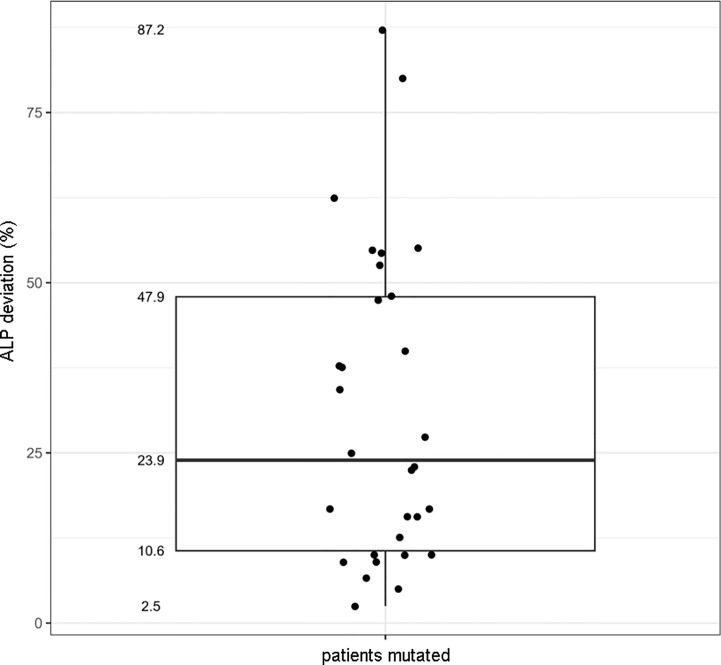
There were 21 ALPL variants and one large genomic deletion found in 20 out of 30 patients. Unexpectedly, NGS-driven differential diagnosis allowed uncovering three hidden additional HPP cases, for a total of 33 HPP subjects. Eight out of 24 coding variants were novel and classified as "pathogenic", "likely pathogenic", and "variants of uncertain significance". Bioinformatic analysis confirmed that all the variants strongly destabilize the homodimer structure. There were 10 cases with low ALP and high VitB6 that resulted negative to genetic testing, whereas two positive cases have an unexpected normal ALP value. No association was evident with other biochemical/clinical parameters.
We present the survey of HPP Italian patients with the highest ALPL mutation rate so far reported and confirm the complexity of a prompt recognition of the syndrome, mostly for HPP in adults. Low ALP and high VitB6 values are mandatory for the genetic screening, this latter remaining the gold standard not only to confirm the clinical diagnosis but also to make differential diagnosis, to identify carriers, to avoid likely dangerous therapy in unrecognized cases.
低磷酸酯酶症(HPP)是一种由 ALPL 基因失活变异引起的罕见遗传性疾病。目前有关意大利 HPP 临床表现的资料有限,也缺乏意大利 HPP 调查的相关数据。
我们从意大利的不同三级医疗机构招募了 30 名疑似 HPP 患者。收集了生物样本及相关临床、生化和病史数据,并对 ALPL 基因进行了测序。还对 ALPL 基因座(1p36)的大片段基因缺失进行了检测。应用系统发育保守性和建模来推断变异对蛋白质结构的影响。
在 30 名患者中的 20 名中发现了 21 种 ALPL 变异和 1 种大片段基因缺失。出乎意料的是,NGS 驱动的鉴别诊断方法发现了另外 3 例隐藏的 HPP 病例,总共 33 例 HPP 患者。24 种编码变异中有 8 种是新的,被归类为“致病性”、“可能致病性”和“意义不明的变异”。生物信息学分析证实,所有变异均强烈破坏了同源二聚体结构。有 10 例 ALP 低和 VitB6 高的患者基因检测结果为阴性,但有 2 例阳性患者的 ALP 值异常正常。未发现与其他生化/临床参数相关。
我们报告了迄今为止意大利 HPP 患者中 ALPL 突变率最高的调查结果,并证实了快速识别该综合征的复杂性,尤其是对于成人 HPP。低 ALP 和高 VitB6 值是遗传筛查的必要条件,基因检测不仅是临床诊断的金标准,也是鉴别诊断、识别携带者、避免在未识别病例中进行可能危险的治疗的必要条件。