Gupta Asmita, Basu Reelina, Bashyam Murali Dharan
Laboratory of Molecular Oncology, Centre of DNA Fingerprinting and Diagnostics, Hyderabad, India.
Access Microbiol. 2023 Jul 20;5(7). doi: 10.1099/acmi.0.000513.v3. eCollection 2023.
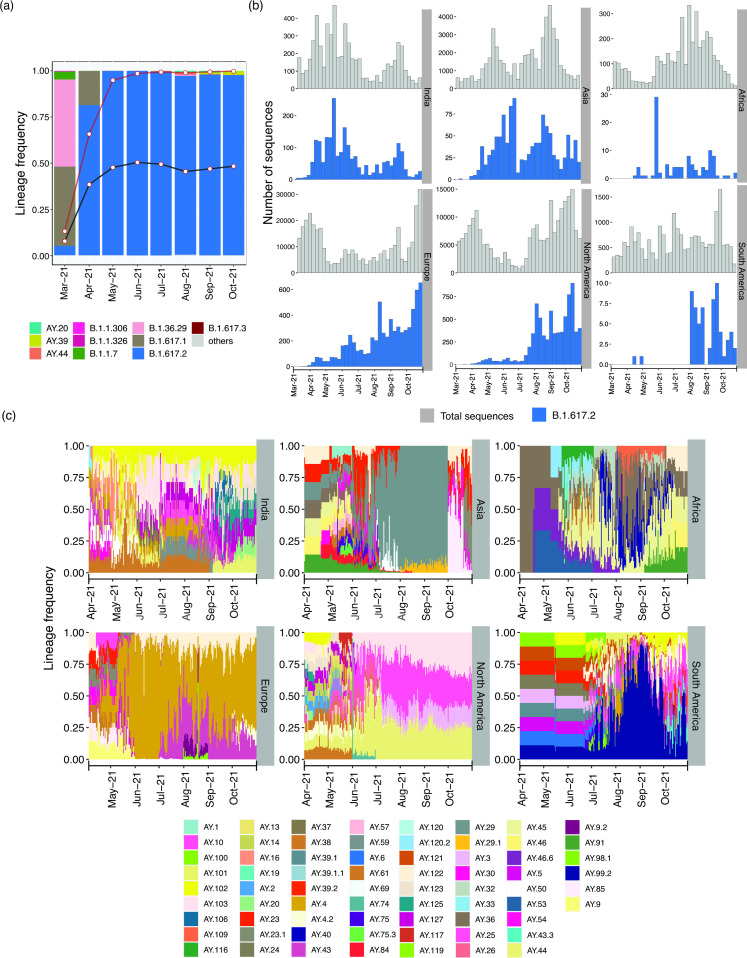
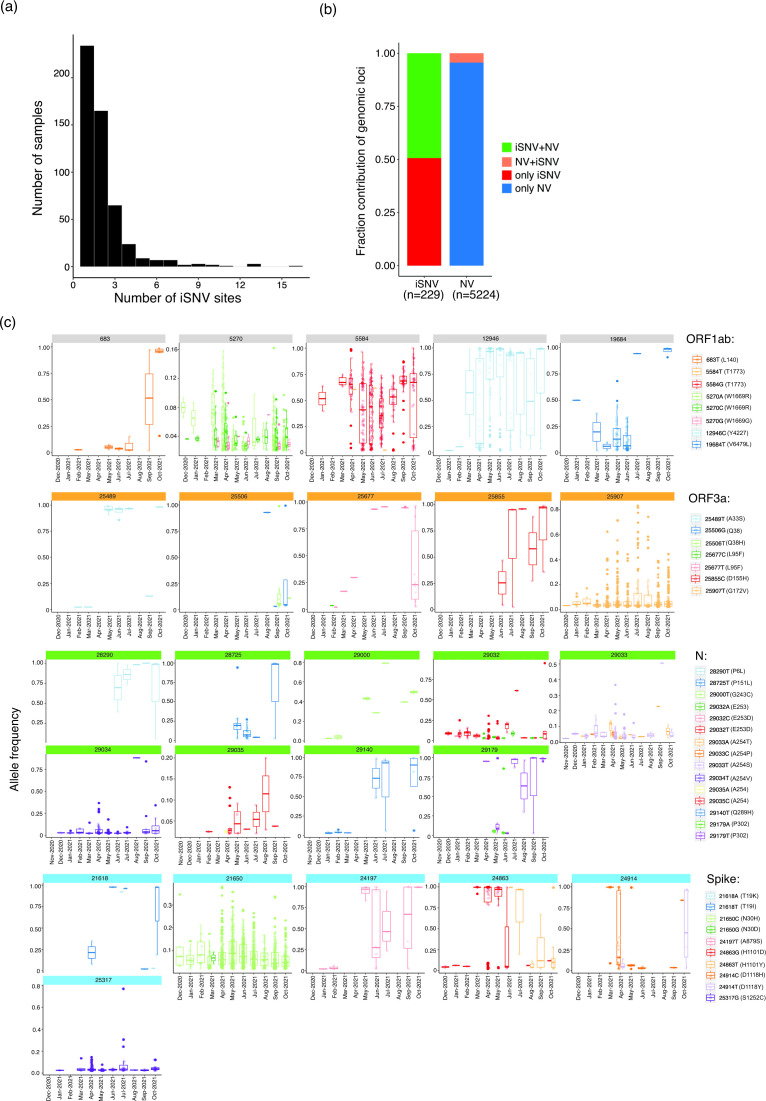
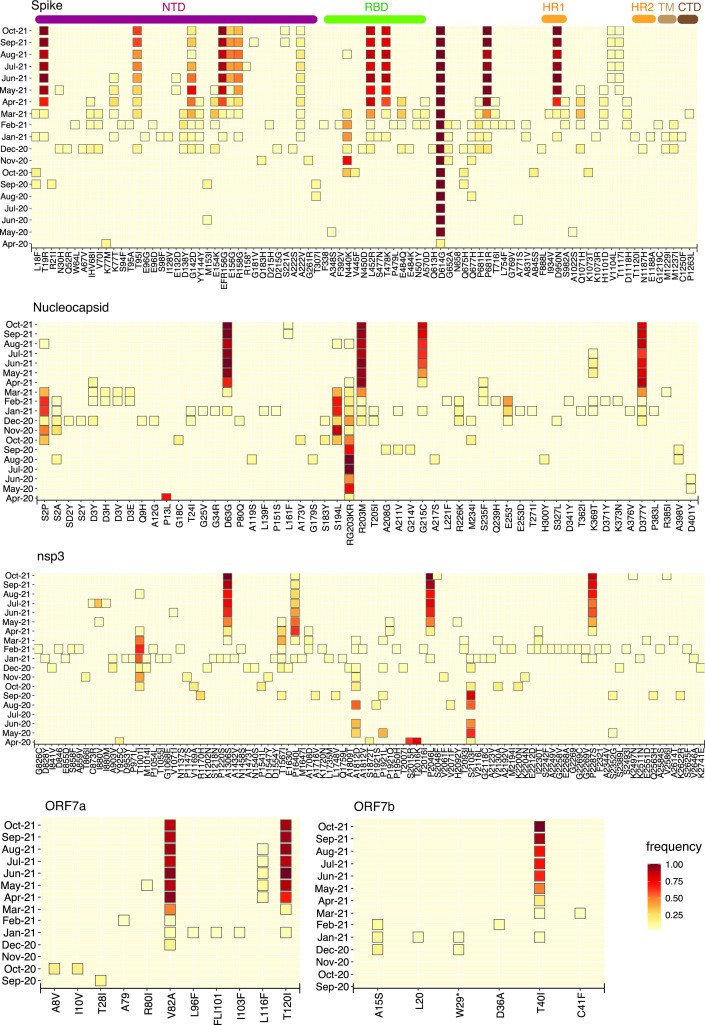
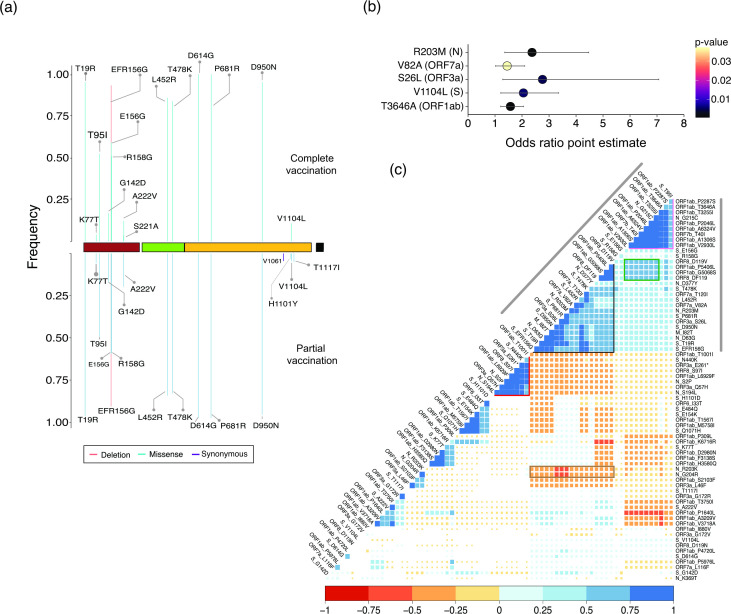
Despite seminal advances towards understanding the infection mechanism of SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2), it continues to cause significant morbidity and mortality worldwide. Though mass immunization programmes have been implemented in several countries, the viral transmission cycle has shown a continuous progression in the form of multiple waves. A constant change in the frequencies of dominant viral lineages, arising from the accumulation of nucleotide variations (NVs) through favourable selection, is understandably expected to be a major determinant of disease severity and possible vaccine escape. Indeed, worldwide efforts have been initiated to identify specific virus lineage(s) and/or NVs that may cause a severe clinical presentation or facilitate vaccination breakthrough. Since host genetics is expected to play a major role in shaping virus evolution, it is imperative to study the role of genome-wide SARS-CoV-2 NVs across various populations. In the current study, we analysed the whole genome sequence of 3543 SARS-CoV-2-infected samples obtained from the state of Telangana, India (including 210 from our previous study), collected over an extended period from April 2020 to October 2021. We present a unique perspective on the evolution of prevalent virus lineages and NVs during this period. We also highlight the presence of specific NVs likely to be associated favourably with samples classified as vaccination breakthroughs. Finally, we report genome-wide intra-host variations at novel genomic positions. The results presented here provide critical insights into virus evolution over an extended period and pave the way to rigorously investigate the role of specific NVs in vaccination breakthroughs.
尽管在理解严重急性呼吸综合征冠状病毒2(SARS-CoV-2)的感染机制方面取得了重大进展,但它仍在全球范围内导致大量发病和死亡。虽然多个国家已实施大规模免疫计划,但病毒传播周期以多波形式持续演进。由于通过有利选择积累核苷酸变异(NVs)导致优势病毒谱系频率不断变化,可以理解,这有望成为疾病严重程度和可能的疫苗逃逸的主要决定因素。事实上,全球已展开努力,以确定可能导致严重临床表现或促成疫苗突破的特定病毒谱系和/或NVs。由于宿主遗传学预计在塑造病毒进化中起主要作用,因此有必要研究全基因组SARS-CoV-2 NVs在不同人群中的作用。在本研究中,我们分析了从印度特伦甘纳邦获得的3543份SARS-CoV-2感染样本的全基因组序列(包括我们先前研究中的210份),这些样本是在2020年4月至2021年10月的较长时间内收集的。我们展示了这一时期流行病毒谱系和NVs进化的独特视角。我们还强调了可能与归类为疫苗突破的样本呈有利关联的特定NVs的存在。最后,我们报告了新基因组位置上的全基因组宿主内变异。此处呈现的结果为长时间的病毒进化提供了关键见解,并为严格研究特定NVs在疫苗突破中的作用铺平了道路。