Brown Brielin C, Wang Collin, Kasela Silva, Aguet François, Nachun Daniel C, Taylor Kent D, Tracy Russell P, Durda Peter, Liu Yongmei, Johnson W Craig, Van Den Berg David, Gupta Namrata, Gabriel Stacy, Smith Joshua D, Gerzsten Robert, Clish Clary, Wong Quenna, Papanicolau George, Blackwell Thomas W, Rotter Jerome I, Rich Stephen S, Barr R Graham, Ardlie Kristin G, Knowles David A, Lappalainen Tuuli
New York Genome Center, New York, NY, USA.
Data Science Institute, Columbia University, New York, NY, USA.
Cell Genom. 2023 Jul 10;3(8):100359. doi: 10.1016/j.xgen.2023.100359. eCollection 2023 Aug 9.
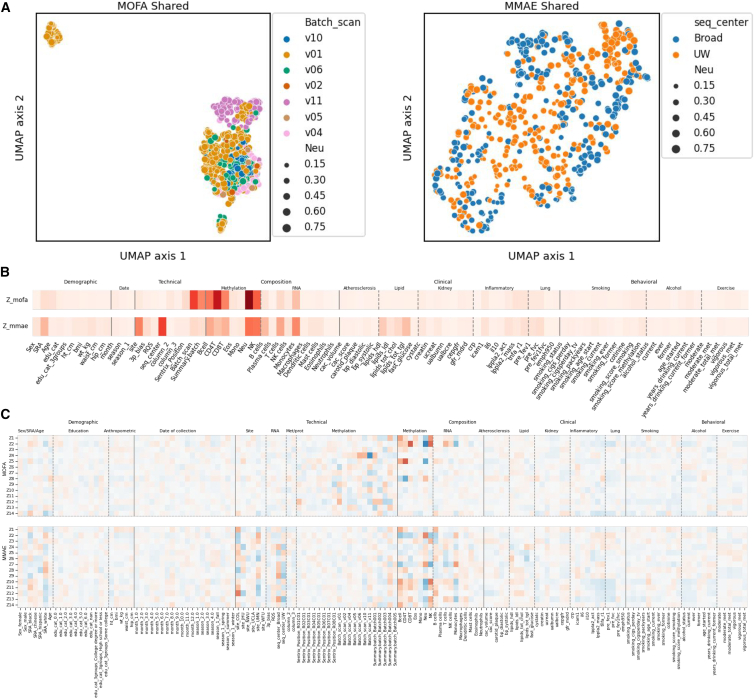
Multi-omics datasets are becoming more common, necessitating better integration methods to realize their revolutionary potential. Here, we introduce multi-set correlation and factor analysis (MCFA), an unsupervised integration method tailored to the unique challenges of high-dimensional genomics data that enables fast inference of shared and private factors. We used MCFA to integrate methylation markers, protein expression, RNA expression, and metabolite levels in 614 diverse samples from the Trans-Omics for Precision Medicine/Multi-Ethnic Study of Atherosclerosis multi-omics pilot. Samples cluster strongly by ancestry in the shared space, even in the absence of genetic information, while private spaces frequently capture dataset-specific technical variation. Finally, we integrated genetic data by conducting a genome-wide association study (GWAS) of our inferred factors, observing that several factors are enriched for GWAS hits and -expression quantitative trait loci. Two of these factors appear to be related to metabolic disease. Our study provides a foundation and framework for further integrative analysis of ever larger multi-modal genomic datasets.
多组学数据集正变得越来越普遍,这就需要更好的整合方法来实现其变革性潜力。在此,我们介绍多集相关性和因子分析(MCFA),这是一种针对高维基因组数据的独特挑战量身定制的无监督整合方法,能够快速推断共享因子和私有因子。我们使用MCFA对精准医学跨组学/动脉粥样硬化多族裔研究多组学试点项目中614个不同样本的甲基化标记、蛋白质表达、RNA表达和代谢物水平进行整合。在共享空间中,样本按祖先强烈聚类,即使在没有遗传信息的情况下也是如此,而私有空间经常捕捉特定于数据集的技术变异。最后,我们通过对推断出的因子进行全基因组关联研究(GWAS)来整合遗传数据,观察到几个因子在GWAS命中和表达数量性状位点方面富集。其中两个因子似乎与代谢疾病有关。我们的研究为进一步整合分析更大规模的多模态基因组数据集提供了基础和框架。