Division of Biostatistics and Bioinformatics, Institute of Population Health Sciences, National Health Research Institutes, No. 35, Keyan Road, Zhunan, 350, Taiwan.
Gigascience. 2019 May 1;8(5). doi: 10.1093/gigascience/giz045.
An integrative multi-omics analysis approach that combines multiple types of omics data including genomics, epigenomics, transcriptomics, proteomics, metabolomics, and microbiomics has become increasing popular for understanding the pathophysiology of complex diseases. Although many multi-omics analysis methods have been developed for complex disease studies, only a few simulation tools that simulate multiple types of omics data and model their relationships with disease status are available, and these tools have their limitations in simulating the multi-omics data.
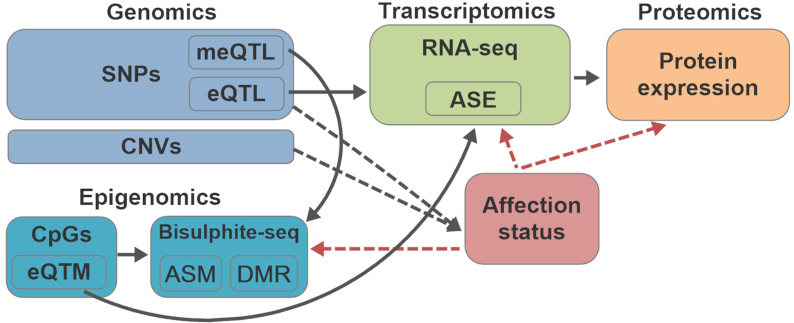
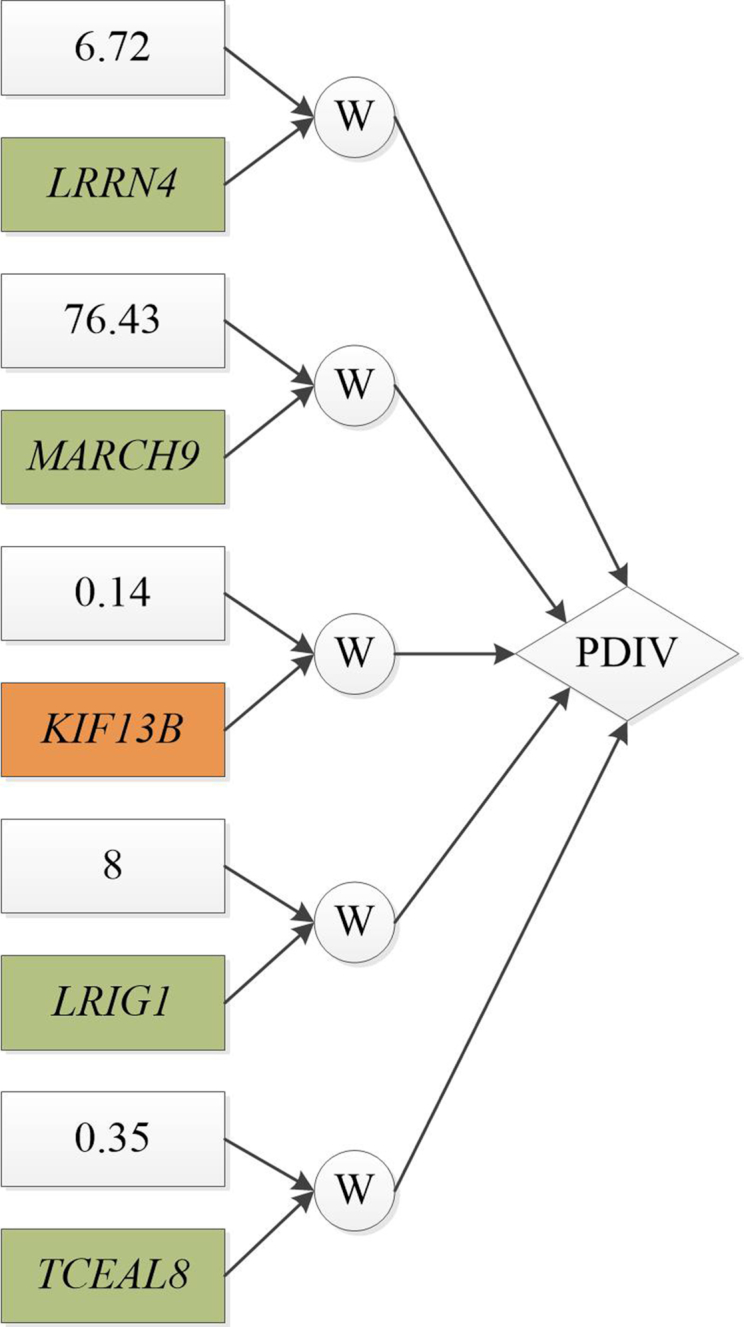
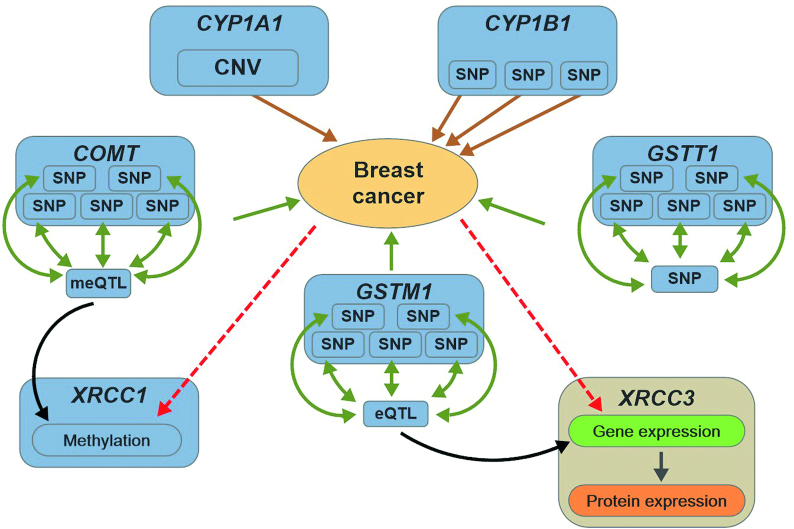
We developed the multi-omics data simulator OmicsSIMLA, which simulates genomics (i.e., single-nucleotide polymorphisms [SNPs] and copy number variations), epigenomics (i.e., bisulphite sequencing), transcriptomics (i.e., RNA sequencing), and proteomics (i.e., normalized reverse phase protein array) data at the whole-genome level. Furthermore, the relationships between different types of omics data, such as methylation quantitative trait loci (SNPs influencing methylation), expression quantitative trait loci (SNPs influencing gene expression), and expression quantitative trait methylations (methylations influencing gene expression), were modeled. More importantly, the relationships between these multi-omics data and the disease status were modeled as well. We used OmicsSIMLA to simulate a multi-omics dataset for breast cancer under a hypothetical disease model and used the data to compare the performance among existing multi-omics analysis methods in terms of disease classification accuracy and runtime. We also used OmicsSIMLA to simulate a multi-omics dataset with a scale similar to an ovarian cancer multi-omics dataset. The neural network-based multi-omics analysis method ATHENA was applied to both the real and simulated data and the results were compared. Our results demonstrated that complex disease mechanisms can be simulated by OmicsSIMLA, and ATHENA showed the highest prediction accuracy when the effects of multi-omics features (e.g., SNPs, copy number variations, and gene expression levels) on the disease were strong. Furthermore, similar results can be obtained from ATHENA when analyzing the simulated and real ovarian multi-omics data.
OmicsSIMLA will be useful to evaluate the performace of different multi-omics analysis methods. Sample sizes and power can also be calculated by OmicsSIMLA when planning a new multi-omics disease study.
综合多组学分析方法越来越受到关注,该方法结合了多种组学数据,包括基因组学、表观基因组学、转录组学、蛋白质组学、代谢组学和微生物组学,用于理解复杂疾病的病理生理学。尽管已经开发了许多用于复杂疾病研究的多组学分析方法,但可用的模拟多种组学数据并模拟它们与疾病状态关系的模拟工具却很少,这些工具在模拟多组学数据方面存在局限性。
我们开发了多组学数据模拟器 OmicsSIMLA,它可以模拟全基因组水平的基因组学(即单核苷酸多态性 [SNP] 和拷贝数变异)、表观基因组学(即亚硫酸氢盐测序)、转录组学(即 RNA 测序)和蛋白质组学(即归一化反相蛋白阵列)数据。此外,还模拟了不同类型的组学数据之间的关系,例如甲基化数量性状基因座(影响甲基化的 SNP)、表达数量性状基因座(影响基因表达的 SNP)和表达数量性状甲基化(影响基因表达的甲基化)。更重要的是,还模拟了这些多组学数据与疾病状态之间的关系。我们使用 OmicsSIMLA 模拟了乳腺癌的多组学数据集,并使用该数据集在疾病分类准确性和运行时方面比较了现有多组学分析方法的性能。我们还使用 OmicsSIMLA 模拟了一个规模与卵巢癌多组学数据集相似的多组学数据集。基于神经网络的多组学分析方法 ATHENA 被应用于真实和模拟数据,并对结果进行了比较。我们的结果表明,OmicsSIMLA 可以模拟复杂的疾病机制,并且当多组学特征(例如 SNP、拷贝数变异和基因表达水平)对疾病的影响较强时,ATHENA 显示出最高的预测准确性。此外,当分析模拟和真实卵巢多组学数据时,ATHENA 可以获得相似的结果。
OmicsSIMLA 将有助于评估不同多组学分析方法的性能。在规划新的多组学疾病研究时,也可以使用 OmicsSIMLA 计算样本量和功效。