Sangster Madison, Shahriar Sanjid, Niziolek Zachary, Carisi Maria Carla, Lewandowski Michael, Budnik Bogdan, Grishchuk Yulia
Center for Genomic Medicine and Department of Neurology, Massachusetts General Hospital Research Institute and Harvard Medical School, Boston, MA, United States.
Wyss Institute for Biologically Inspired Engineering, Harvard University, Boston, MA, United States.
Front Mol Neurosci. 2023 Aug 7;16:1215425. doi: 10.3389/fnmol.2023.1215425. eCollection 2023.
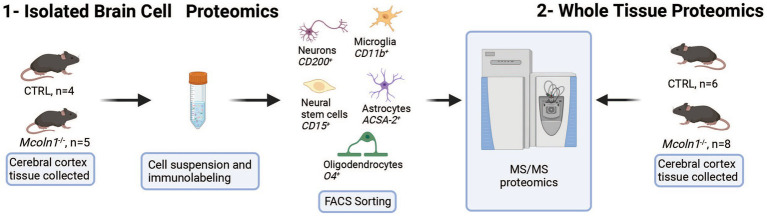
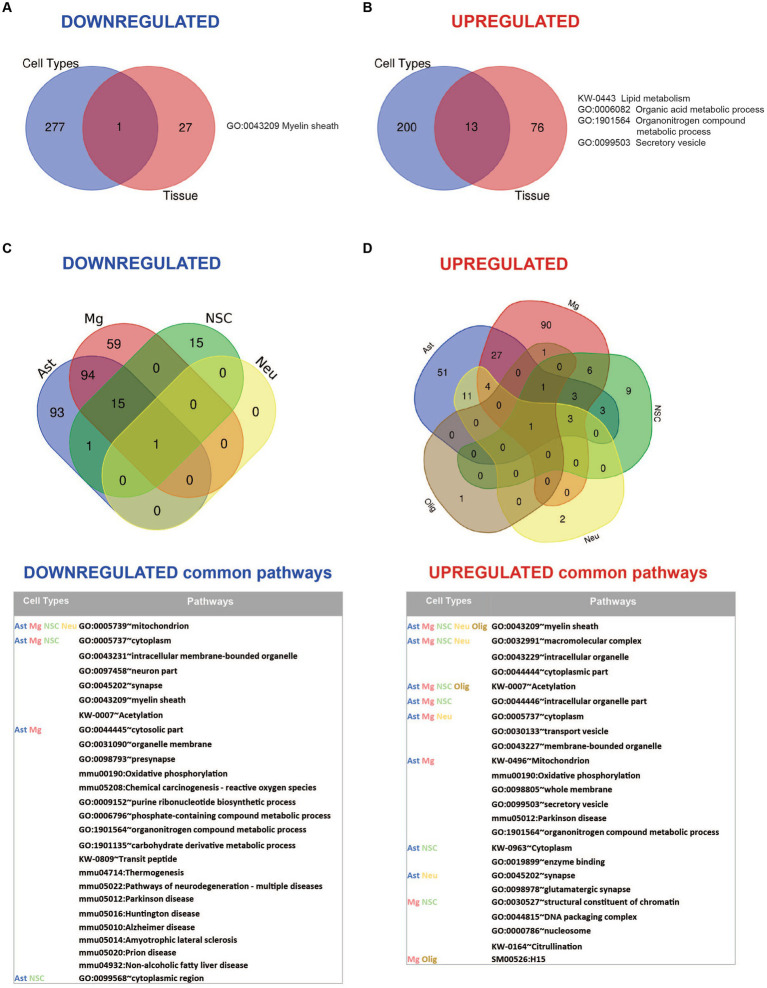
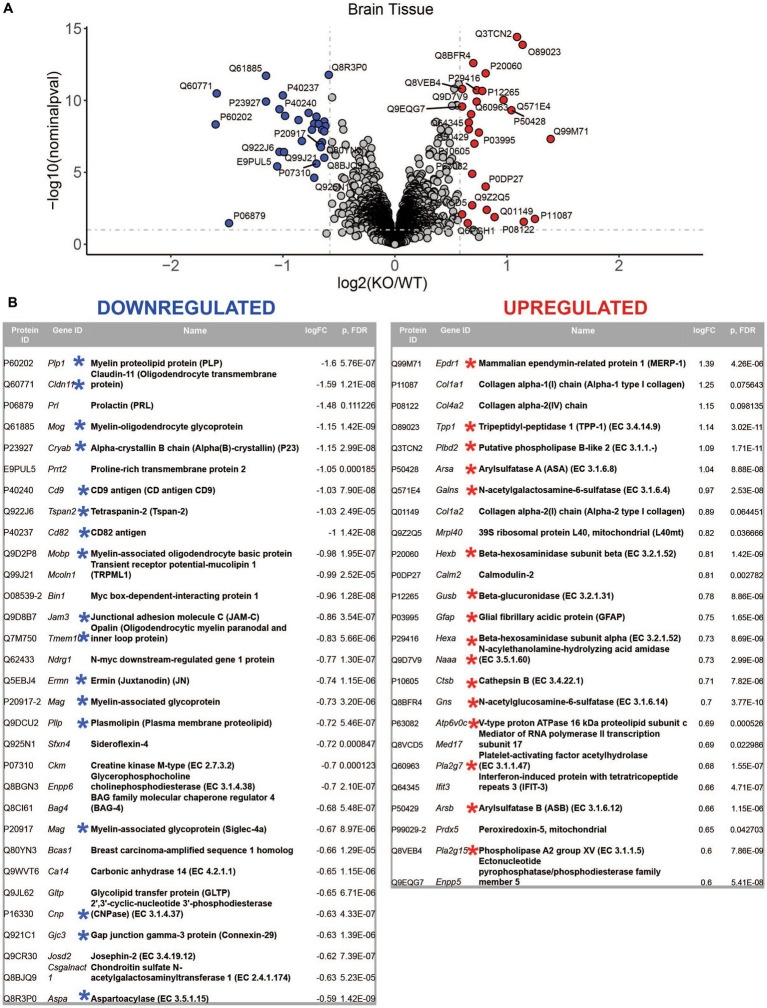
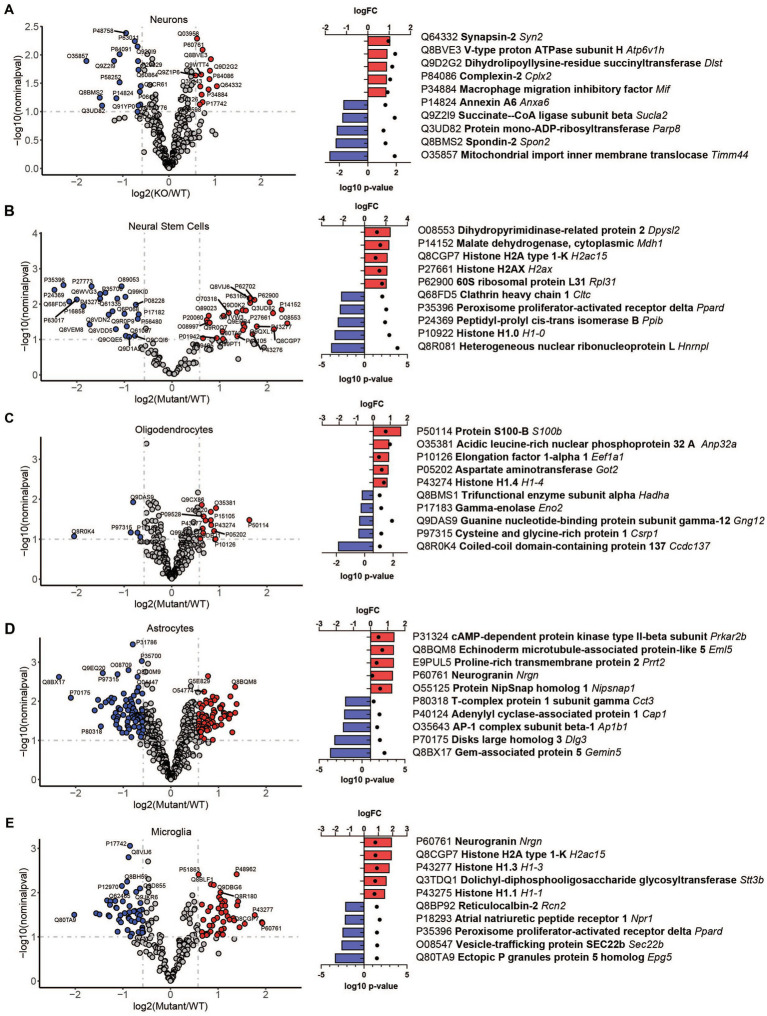
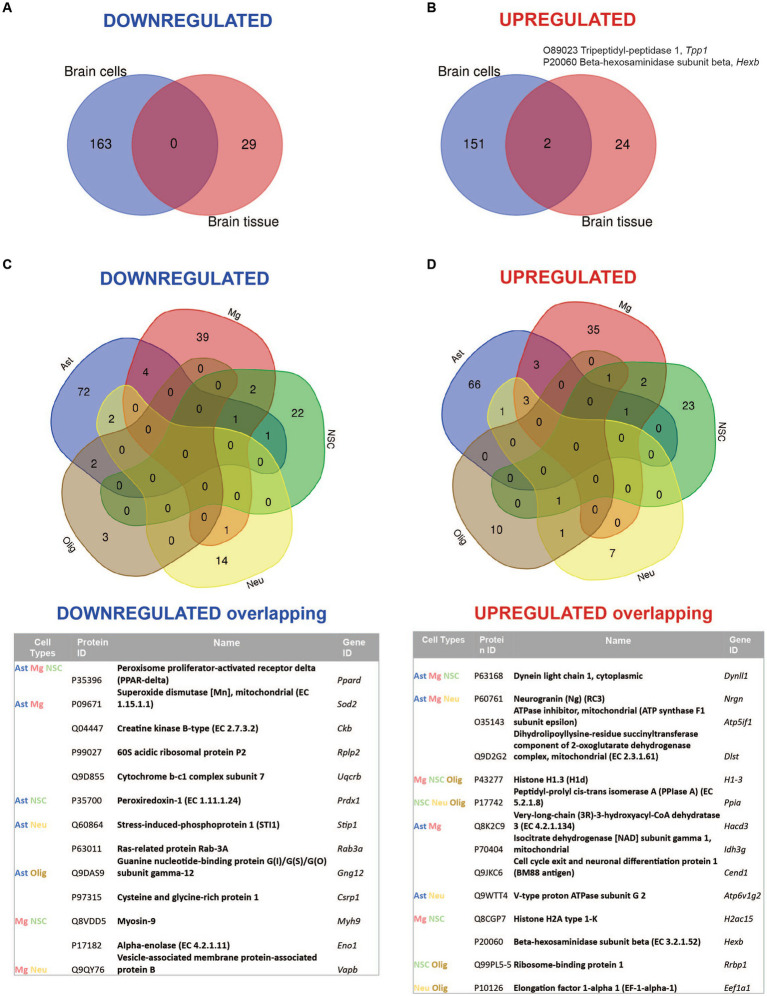
Mucolipidosis IV (MLIV) is an ultra-rare, recessively inherited lysosomal disorder resulting from inactivating mutations in , the gene encoding the lysosomal cation channel TRPML1. The disease primarily affects the central nervous system (CNS) and manifests in the first year with cognitive and motor developmental delay, followed by a gradual decline in neurological function across the second decade of life, blindness, and premature death in third or fourth decades. Brain pathology manifestations in MLIV are consistent with hypomyelinating leukodystrophy with brain iron accumulation. Presently, there are no approved or investigational therapies for MLIV, and pathogenic mechanisms remain largely unknown. The MLIV mouse model, mice, recapitulates all major manifestations of the human disease. Here, to better understand the pathological mechanisms in the MLIV brain, we performed cell type specific LC-MS/MS proteomics analysis in the MLIV mouse model and reconstituted molecular signatures of the disease in either freshly isolated populations of neurons, astrocytes, oligodendrocytes, and neural stem cells, or whole tissue cortical homogenates from young adult symptomatic mice. Our analysis confirmed on the molecular level major histopathological hallmarks of MLIV universally present in tissue and brain cells, such as hypomyelination, lysosomal dysregulation, and impaired metabolism of lipids and polysaccharides. Importantly, pathway analysis in brain cells revealed mitochondria-related alterations in all brain cells, except oligodendrocytes, that was not possible to resolve in whole tissue. We also report unique proteome signatures and dysregulated pathways for each brain cell population used in this study. These data shed new light on cell-intrinsic mechanisms of MLIV and provide new insights for biomarker discovery and validation to advance translational studies for this disease.
黏脂贮积症IV型(MLIV)是一种极其罕见的隐性遗传溶酶体疾病,由编码溶酶体阳离子通道TRPML1的基因发生失活突变所致。该疾病主要影响中枢神经系统(CNS),在出生后第一年表现为认知和运动发育迟缓,随后在第二个十年中神经功能逐渐衰退,出现失明,并在第三或第四个十年中过早死亡。MLIV的脑部病理表现与伴有脑铁蓄积的低髓鞘性脑白质营养不良一致。目前,尚无针对MLIV的获批或正在研究的疗法,其致病机制在很大程度上仍不清楚。MLIV小鼠模型,即小鼠,重现了人类疾病的所有主要表现。在此,为了更好地理解MLIV脑内的病理机制,我们在MLIV小鼠模型中进行了细胞类型特异性的液相色谱-串联质谱(LC-MS/MS)蛋白质组学分析,并在新鲜分离的神经元、星形胶质细胞、少突胶质细胞和神经干细胞群体或来自年轻成年有症状小鼠的全脑组织皮质匀浆中重构了该疾病的分子特征。我们的分析在分子水平上证实了MLIV在组织和脑细胞中普遍存在的主要组织病理学特征,如低髓鞘化、溶酶体失调以及脂质和多糖代谢受损。重要的是,对脑细胞的通路分析揭示了除少突胶质细胞外所有MLIV脑细胞中线粒体相关的改变,而这在全脑组织中无法分辨。我们还报告了本研究中使用的每个脑细胞群体独特的蛋白质组特征和失调的通路。这些数据为MLIV的细胞内在机制提供了新的线索,并为生物标志物的发现和验证提供了新的见解,以推动该疾病的转化研究。