Department of Microbiology, School of Basic Medicine, Air Force Medical University, Xi'an, 710032, China.
Department of Neurology, Xijing Hospital, Air Force Medical University, Xi'an, 710032, China; Center of Clinical Aerospace Medicine, School of Aerospace Medicine, Key Laboratory of Aerospace Medicine of Ministry of Education, Air Force Medical University, Xi'an, 710032, China.
Virol Sin. 2023 Oct;38(5):741-754. doi: 10.1016/j.virs.2023.08.006. Epub 2023 Aug 25.
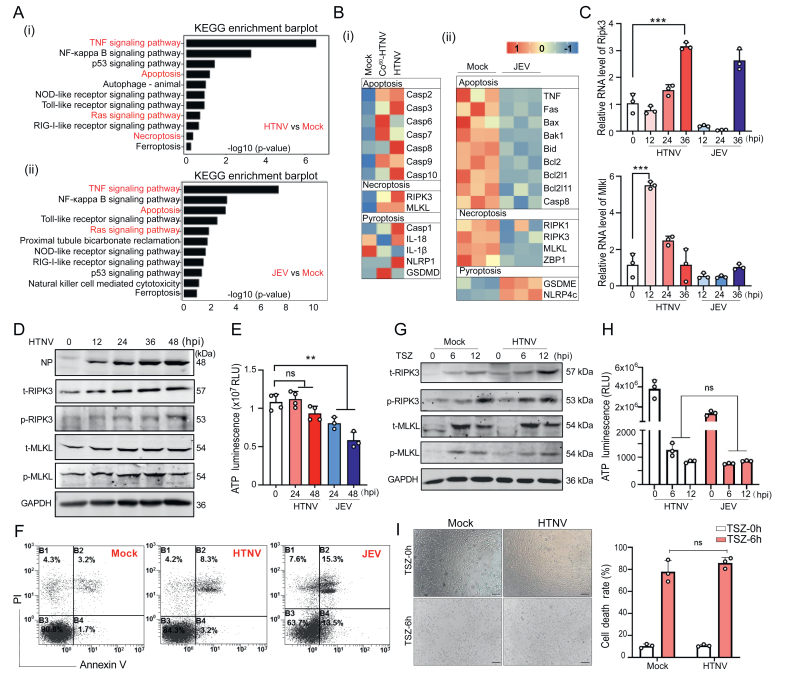
Hantaan virus (HTNV) is a rodent-borne virus that causes hemorrhagic fever with renal syndrome (HFRS), resulting in a high mortality rate of 15%. Interferons (IFNs) play a critical role in the anti-hantaviral immune response, and IFN pretreatment efficiently restricts HTNV infection by triggering the expression of a series of IFN-stimulated genes (ISGs) through the Janus kinase-signal transducer and activator of transcription 1 (JAK-STAT) pathway. However, the tremendous amount of IFNs produced during late infection could not restrain HTNV replication, and the mechanism remains unclear. Here, we demonstrated that receptor-interacting protein kinase 3 (RIPK3), a crucial molecule that mediates necroptosis, was activated by HTNV and contributed to hantavirus evasion of IFN responses by inhibiting STAT1 phosphorylation. RNA-seq analysis revealed the upregulation of multiple cell death-related genes after HTNV infection, with RIPK3 identified as a key modulator of viral replication. RIPK3 ablation significantly enhanced ISGs expression and restrained HTNV replication, without affecting the expression of pattern recognition receptors (PRRs) or the production of type I IFNs. Conversely, exogenously expressed RIPK3 compromised the host's antiviral response and facilitated HTNV replication. RIPK3 mice also maintained a robust ability to clear HTNV with enhanced innate immune responses. Mechanistically, we found that RIPK3 could bind STAT1 and inhibit STAT1 phosphorylation dependent on the protein kinase domain (PKD) of RIPK3 but not its kinase activity. Overall, these observations demonstrated a noncanonical function of RIPK3 during viral infection and have elucidated a novel host innate immunity evasion strategy utilized by HTNV.
汉坦病毒(HTNV)是一种啮齿动物传播的病毒,可引起肾综合征出血热(HFRS),导致 15%的高死亡率。干扰素(IFNs)在抗汉坦病毒免疫反应中发挥着关键作用,IFN 预处理通过 JAK-STAT 途径触发一系列干扰素刺激基因(ISGs)的表达,从而有效地限制 HTNV 感染。然而,在晚期感染期间产生的大量 IFN 无法抑制 HTNV 的复制,其机制尚不清楚。在这里,我们证明 HTNV 激活了受体相互作用蛋白激酶 3(RIPK3),这是一种介导坏死性凋亡的关键分子,通过抑制 STAT1 磷酸化,促进汉坦病毒逃避 IFN 反应。RNA-seq 分析显示,HTNV 感染后多个细胞死亡相关基因上调,其中 RIPK3 被鉴定为病毒复制的关键调节剂。RIPK3 缺失显著增强了 ISGs 的表达并抑制了 HTNV 的复制,而不影响模式识别受体(PRRs)的表达或 I 型 IFN 的产生。相反,外源性表达的 RIPK3 削弱了宿主的抗病毒反应并促进了 HTNV 的复制。RIPK3 小鼠也保持了强大的清除 HTNV 的能力,并增强了先天免疫反应。在机制上,我们发现 RIPK3 可以结合 STAT1 并抑制依赖 RIPK3 的蛋白激酶结构域(PKD)但不依赖其激酶活性的 STAT1 磷酸化。总的来说,这些观察结果表明 RIPK3 在病毒感染过程中具有非典型功能,并阐明了 HTNV 利用的一种新的宿主先天免疫逃避策略。