Department of Neurology and Neurosurgery, McGill University, Montréal, QC H3A 1A1, Canada.
Child Health and Human Development Program, Research Institute of the McGill University Health Centre, Montréal, QC H4A 3J1, Canada.
Brain. 2023 Dec 1;146(12):5070-5085. doi: 10.1093/brain/awad249.
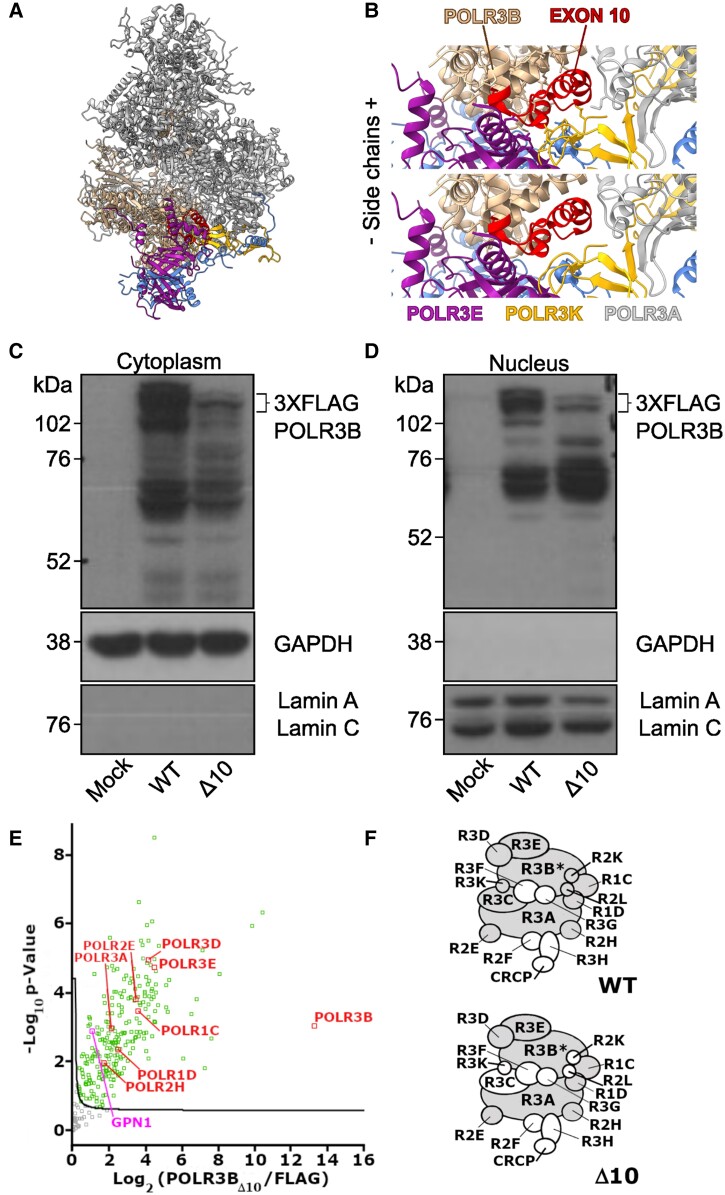
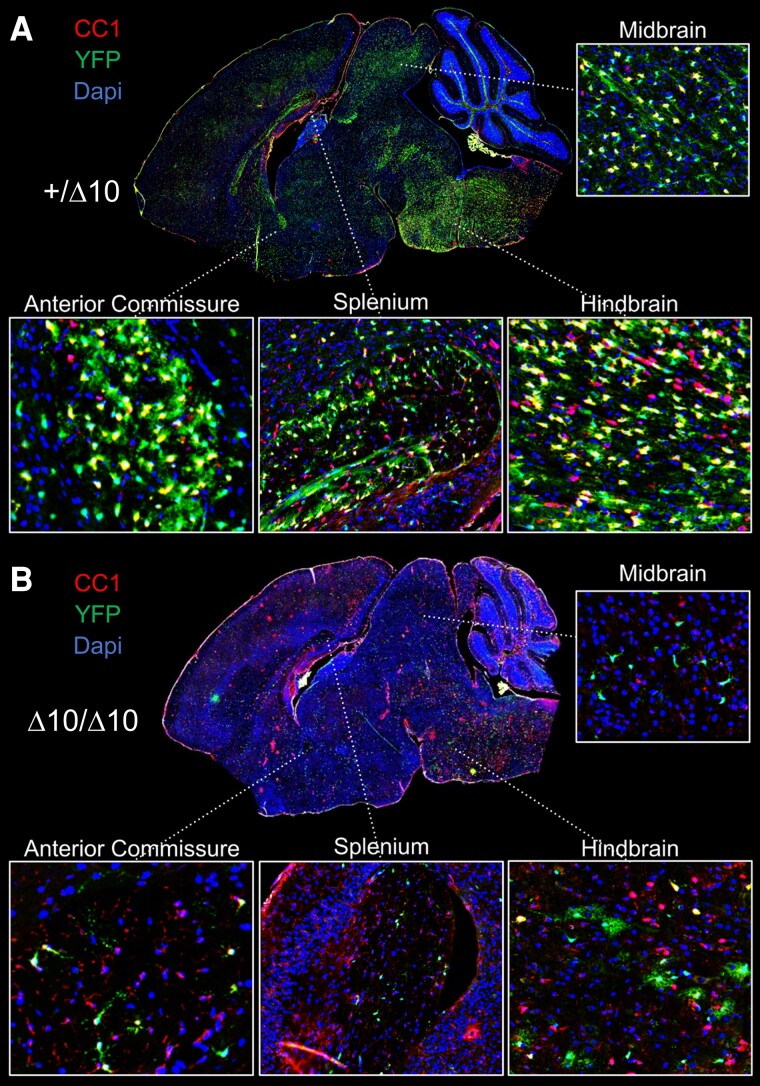
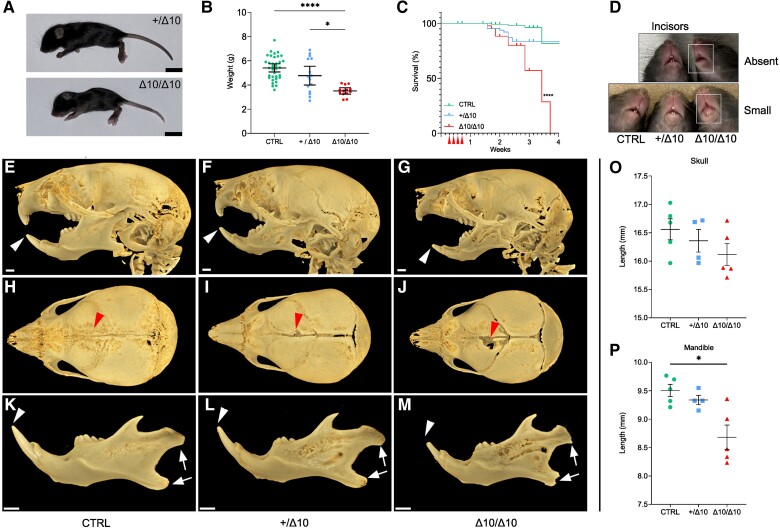
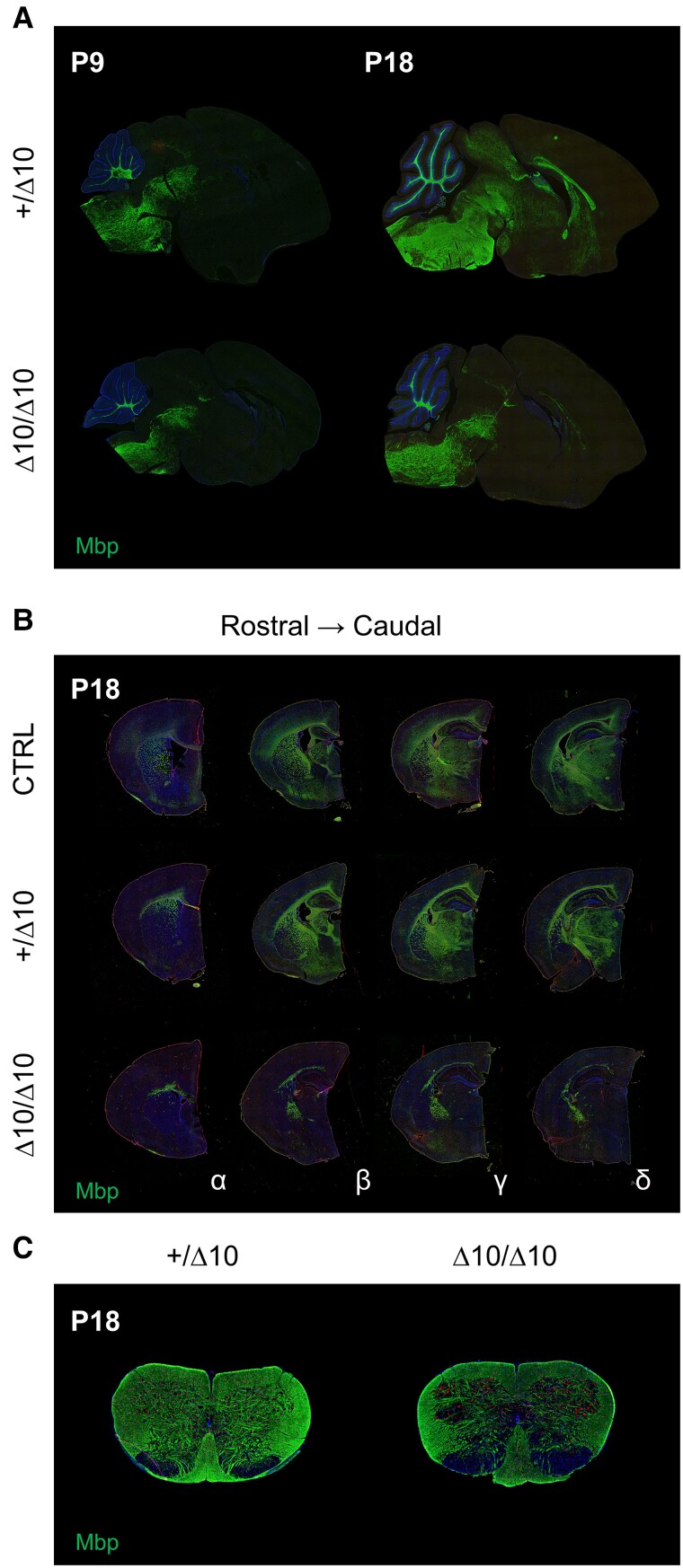
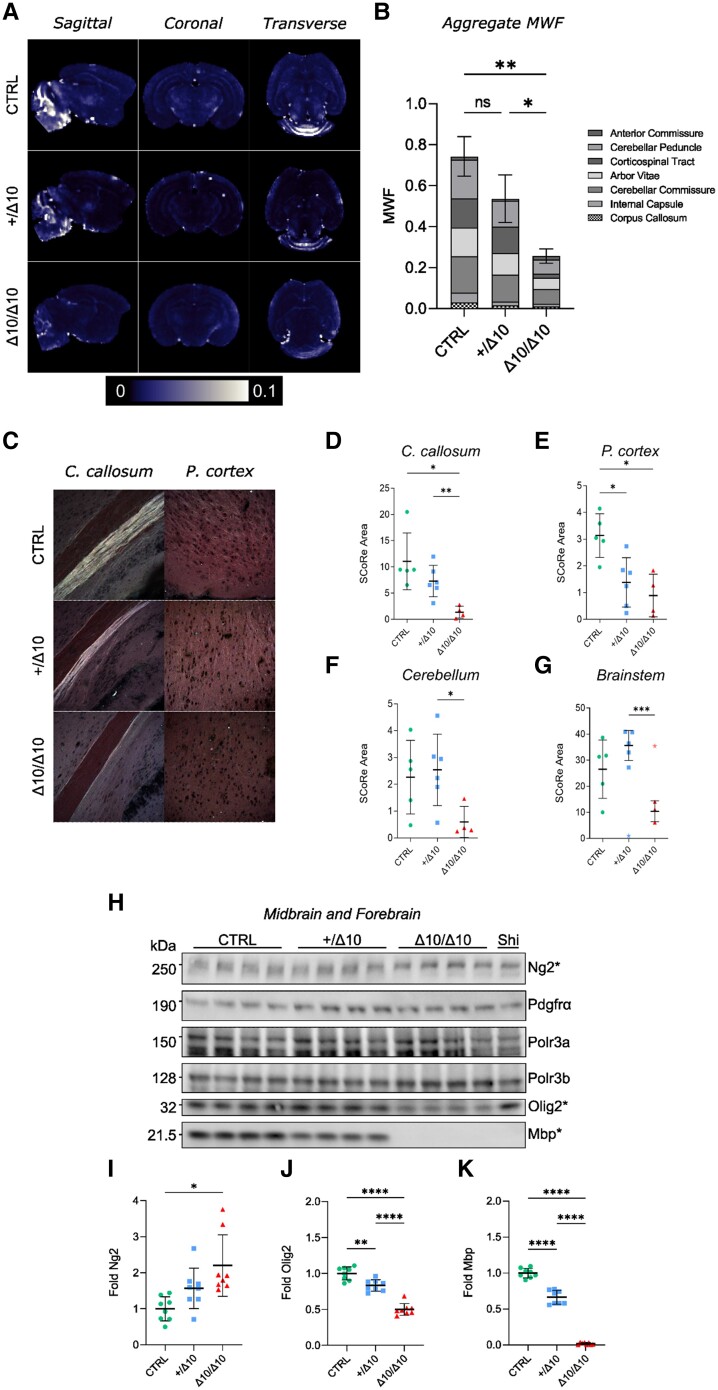
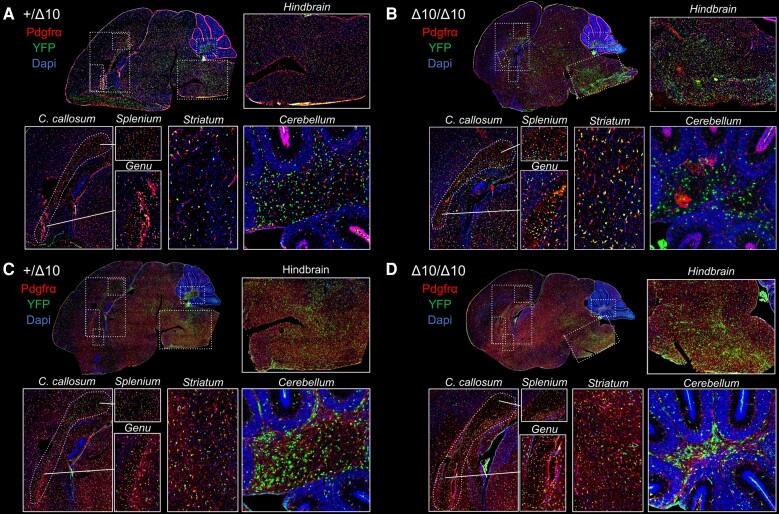
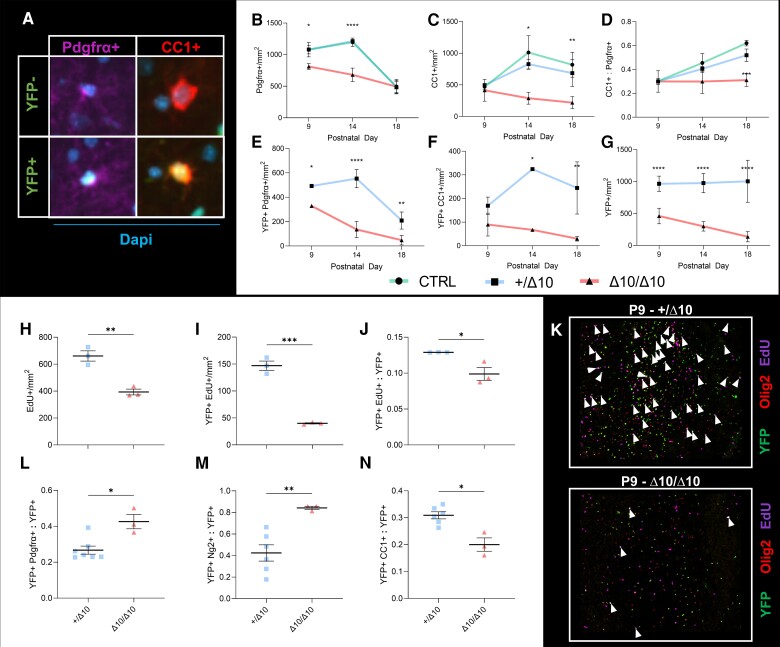
RNA polymerase III (Pol III)-related hypomyelinating leukodystrophy (POLR3-HLD), also known as 4H leukodystrophy, is a severe neurodegenerative disease characterized by the cardinal features of hypomyelination, hypodontia and hypogonadotropic hypogonadism. POLR3-HLD is caused by biallelic pathogenic variants in genes encoding Pol III subunits. While approximately half of all patients carry mutations in POLR3B encoding the RNA polymerase III subunit B, there is no in vivo model of leukodystrophy based on mutation of this Pol III subunit. Here, we determined the impact of POLR3BΔ10 (Δ10) on Pol III in human cells and developed and characterized an inducible/conditional mouse model of leukodystrophy using the orthologous Δ10 mutation in mice. The molecular mechanism of Pol III dysfunction was determined in human cells by affinity purification-mass spectrometry and western blot. Postnatal induction with tamoxifen induced expression of the orthologous Δ10 hypomorph in triple transgenic Pdgfrα-Cre/ERT; R26-Stopfl-EYFP; Polr3bfl mice. CNS and non-CNS features were characterized using a variety of techniques including microCT, ex vivo MRI, immunofluorescence, immunohistochemistry, spectral confocal reflectance microscopy and western blot. Lineage tracing and time series analysis of oligodendrocyte subpopulation dynamics based on co-labelling with lineage-specific and/or proliferation markers were performed. Proteomics suggested that Δ10 causes a Pol III assembly defect, while western blots demonstrated reduced POLR3BΔ10 expression in the cytoplasm and nucleus in human cells. In mice, postnatal Pdgfrα-dependent expression of the orthologous murine mutant protein resulted in recessive phenotypes including severe hypomyelination leading to ataxia, tremor, seizures and limited survival, as well as hypodontia and craniofacial abnormalities. Hypomyelination was confirmed and characterized using classic methods to quantify myelin components such as myelin basic protein and lipids, results which agreed with those produced using modern methods to quantify myelin based on the physical properties of myelin membranes. Lineage tracing uncovered the underlying mechanism for the hypomyelinating phenotype: defective oligodendrocyte precursor proliferation and differentiation resulted in a failure to produce an adequate number of mature oligodendrocytes during postnatal myelinogenesis. In summary, we characterized the Polr3bΔ10 mutation and developed an animal model that recapitulates features of POLR3-HLD caused by POLR3B mutations, shedding light on disease pathogenesis, and opening the door to the development of therapeutic interventions.
RNA 聚合酶 III(Pol III)相关的脑白质营养不良(POLR3-HLD),也称为 4H 脑白质营养不良,是一种严重的神经退行性疾病,其特征为少突胶质细胞形成不良、牙缺失和促性腺激素低下性性腺功能减退症。POLR3-HLD 是由编码 Pol III 亚基的基因中的双等位致病性变异引起的。虽然大约一半的患者携带编码 RNA 聚合酶 III 亚基 B 的 POLR3B 基因突变,但目前尚无基于该 Pol III 亚基突变的脑白质营养不良的体内模型。在这里,我们确定了 POLR3BΔ10(Δ10)对人类细胞中 Pol III 的影响,并使用小鼠中的同源 Δ10 突变开发和表征了一种可诱导/条件性脑白质营养不良动物模型。通过亲和纯化-质谱和 Western blot 确定了 Pol III 功能障碍的分子机制。用他莫昔芬进行出生后诱导,可在三重转基因 Pdgfrα-Cre/ERT;R26-Stopfl-EYFP;Polr3bfl 小鼠中诱导表达同源 Δ10 低聚物。使用各种技术包括 microCT、离体 MRI、免疫荧光、免疫组化、光谱共焦反射率显微镜和 Western blot 来描述 CNS 和非 CNS 特征。基于谱系特异性和/或增殖标志物的共标记进行少突胶质细胞亚群动态的谱系追踪和时间序列分析。蛋白质组学表明 Δ10 导致 Pol III 组装缺陷,而 Western blot 表明人类细胞中细胞质和核内的 POLR3BΔ10 表达减少。在小鼠中,Pdgfrα 依赖性表达同源突变鼠蛋白导致隐性表型,包括严重的少突胶质细胞形成不良导致共济失调、震颤、癫痫发作和有限的存活,以及牙缺失和颅面异常。使用经典方法定量髓鞘成分(如髓鞘碱性蛋白和脂质)来确认和表征少突胶质细胞形成不良,结果与使用基于髓鞘膜物理特性来定量髓鞘的现代方法一致。谱系追踪揭示了少突胶质细胞形成不良表型的潜在机制:少突胶质前体细胞增殖和分化缺陷导致在出生后髓鞘发生期间无法产生足够数量的成熟少突胶质细胞。总之,我们对 Polr3bΔ10 突变进行了表征,并开发了一种动物模型,该模型可再现由 POLR3B 突变引起的 POLR3-HLD 的特征,揭示了疾病发病机制,并为治疗干预措施的开发开辟了道路。