Institute of Translational Medicine, Center for Precision Genome Editing and Genetic Technologies for Biomedicine, Pirogov Russian National Research Medical University, Moscow, Russia.
Laboratory of Structural Bioinformatics, Institute of Biomedical Chemistry, Moscow, Russia.
Front Immunol. 2023 Aug 15;14:1224969. doi: 10.3389/fimmu.2023.1224969. eCollection 2023.
T-cell receptor (TCR) recognition of foreign peptides presented by the major histocompatibility complex (MHC) initiates the adaptive immune response against pathogens. While a large number of TCR sequences specific to different antigenic peptides are known to date, the structural data describing the conformation and contacting residues for TCR-peptide-MHC complexes is relatively limited. In the present study we aim to extend and analyze the set of available structures by performing highly accurate template-based modeling of these complexes using TCR sequences with known specificity.
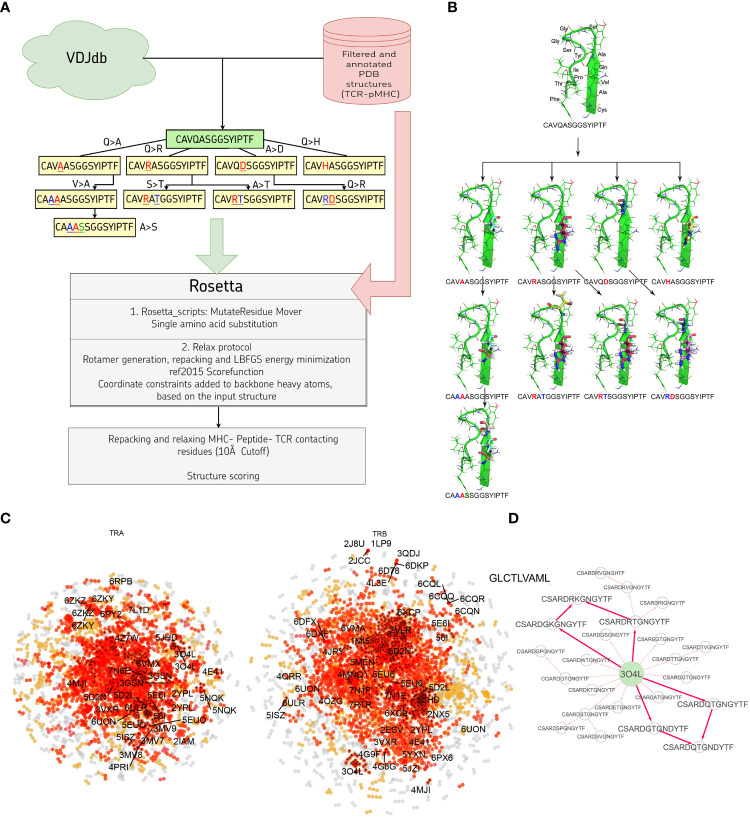
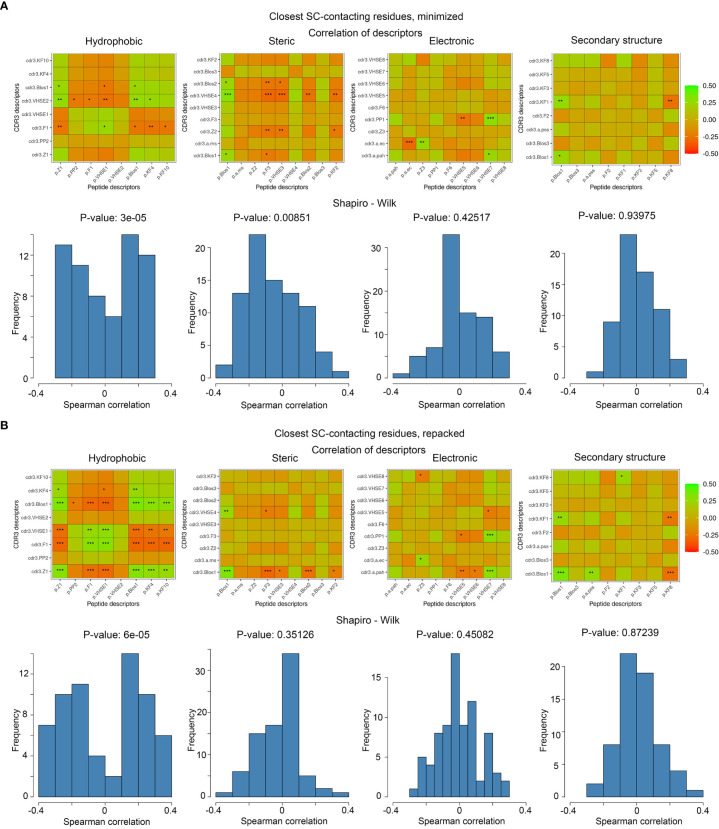
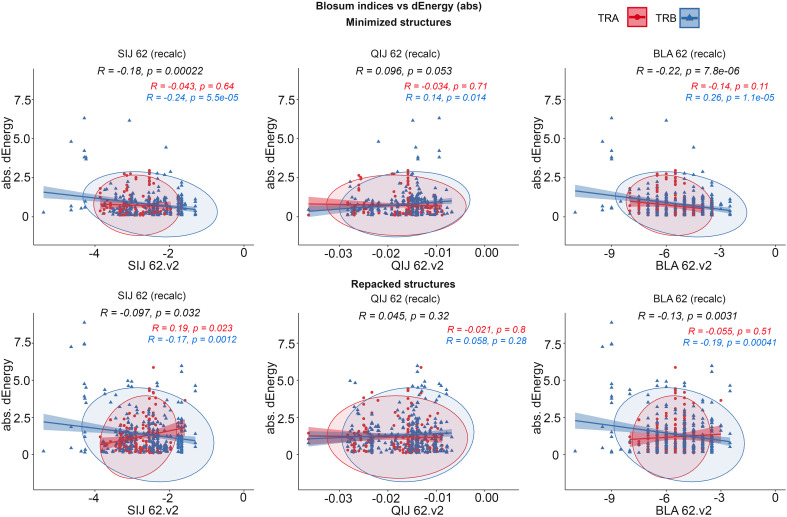
Identification of CDR3 sequences and their further clustering, based on available spatial structures, V- and J-genes of corresponding T-cell receptors, and epitopes, was performed using the VDJdb database. Modeling of the selected CDR3 loops was conducted using a stepwise introduction of single amino acid substitutions to the template PDB structures, followed by optimization of the TCR-peptide-MHC contacting interface using the Rosetta package applications. Statistical analysis and recursive feature elimination procedures were carried out on computed energy values and properties of contacting amino acid residues between CDR3 loops and peptides, using R.
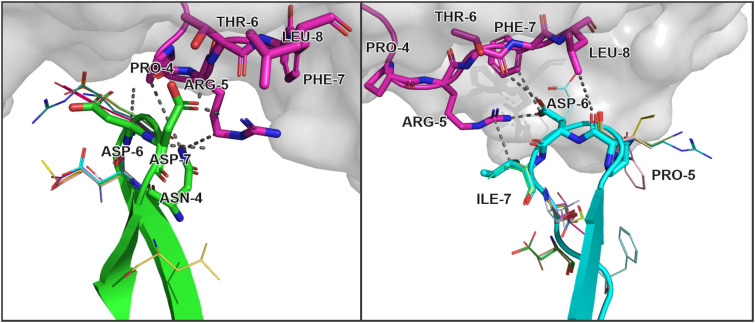
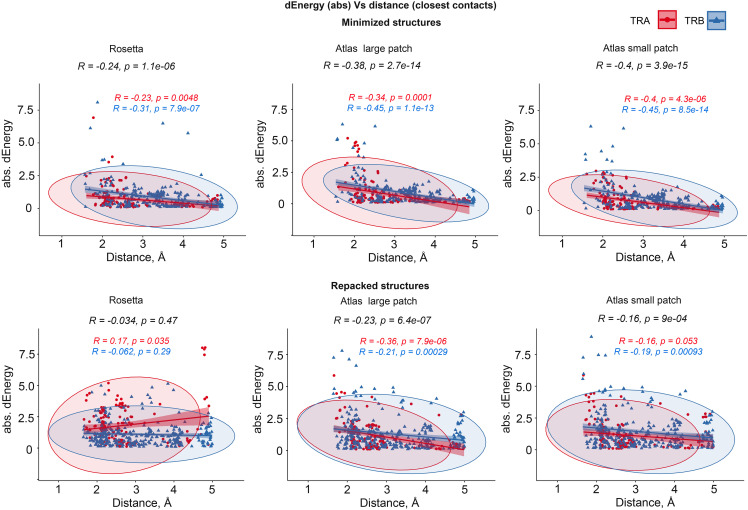
Using the set of 29 complex templates (including a template with SARS-CoV-2 antigen) and 732 specificity records, we built a database of 1585 model structures carrying substitutions in either TCRα or TCRβ chains with some models representing the result of different mutation pathways for the same final structure. This database allowed us to analyze features of amino acid contacts in TCR - peptide interfaces that govern antigen recognition preferences and interpret these interactions in terms of physicochemical properties of interacting residues.
Our results provide a methodology for creating high-quality TCR-peptide-MHC models for antigens of interest that can be utilized to predict TCR specificity.
T 细胞受体 (TCR) 识别主要组织相容性复合体 (MHC) 呈现的外来肽,引发针对病原体的适应性免疫反应。虽然迄今为止已经知道了大量针对不同抗原肽的 TCR 序列,但描述 TCR-肽-MHC 复合物构象和接触残基的结构数据相对有限。在本研究中,我们旨在通过使用具有已知特异性的 TCR 序列对这些复合物进行高度准确的基于模板的建模来扩展和分析现有结构集。
使用 VDJdb 数据库,根据可用的空间结构、相应 T 细胞受体的 V 和 J 基因以及表位,对 CDR3 序列进行鉴定和进一步聚类。使用逐步引入到模板 PDB 结构中的单个氨基酸取代来对所选 CDR3 环进行建模,然后使用 Rosetta 包应用程序优化 TCR-肽-MHC 接触界面。使用 R 对计算出的能量值和 CDR3 环与肽之间的接触氨基酸残基的特性进行统计分析和递归特征消除过程。
使用 29 个复合物模板(包括 SARS-CoV-2 抗原模板)和 732 个特异性记录,我们构建了一个包含 TCRα 或 TCRβ 链取代的 1585 个模型结构的数据库,其中一些模型代表了同一最终结构的不同突变途径的结果。该数据库使我们能够分析 TCR-肽界面中氨基酸接触的特征,这些特征决定了抗原识别偏好,并根据相互作用残基的物理化学性质解释这些相互作用。
我们的结果提供了一种用于为感兴趣的抗原创建高质量 TCR-肽-MHC 模型的方法,可用于预测 TCR 特异性。