Ubben Center for Pulmonary Fibrosis Research, Division of Pulmonary, Critical Care and Sleep Medicine, Department of Internal Medicine, Morsani College of Medicine, University of South Florida, Tampa, Florida, United States.
Am J Physiol Cell Physiol. 2023 Oct 1;325(4):C1046-C1057. doi: 10.1152/ajpcell.00302.2023. Epub 2023 Sep 11.
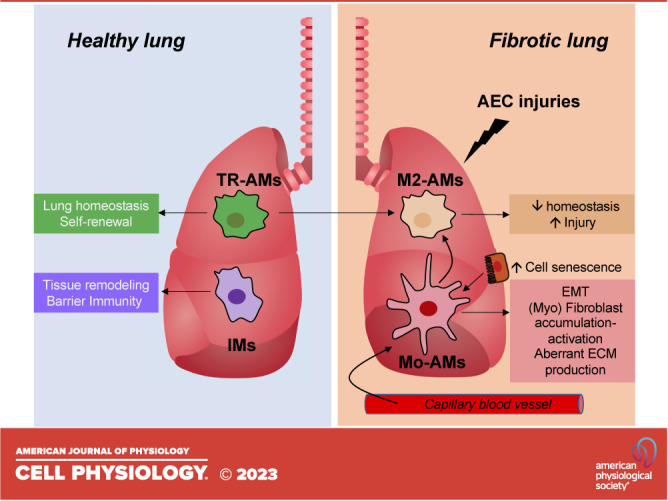
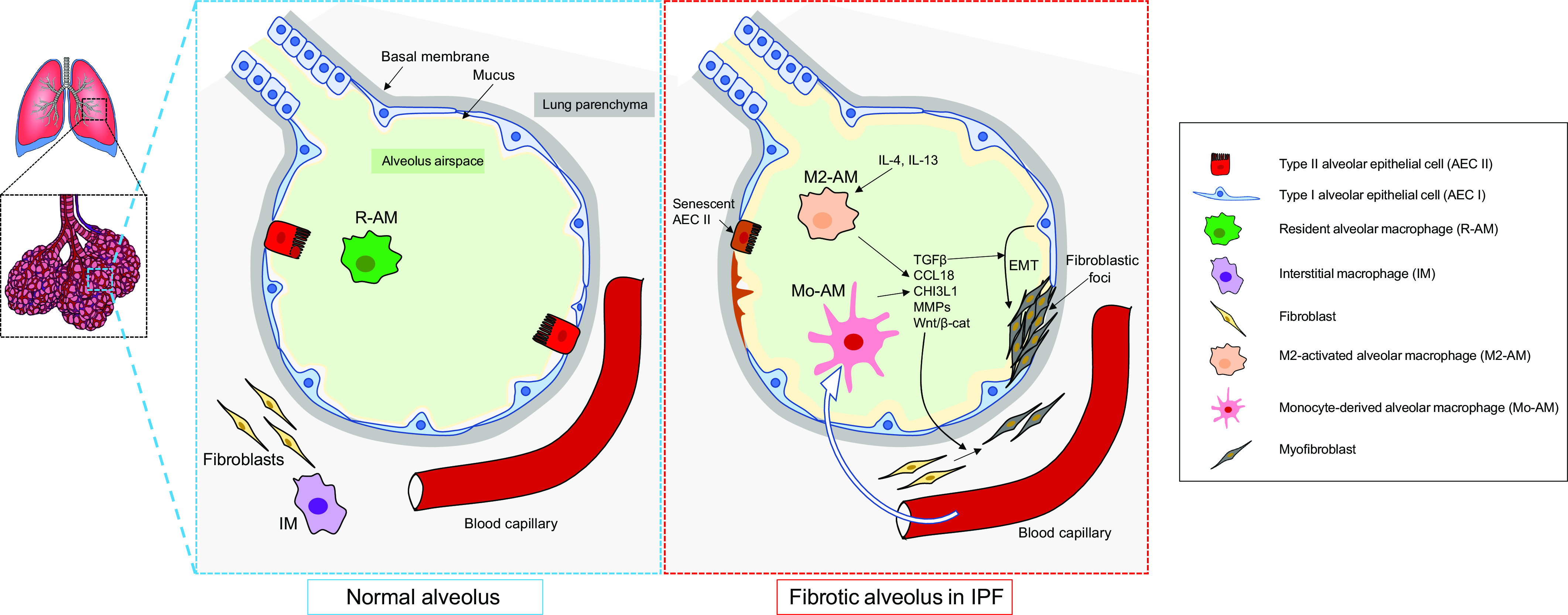
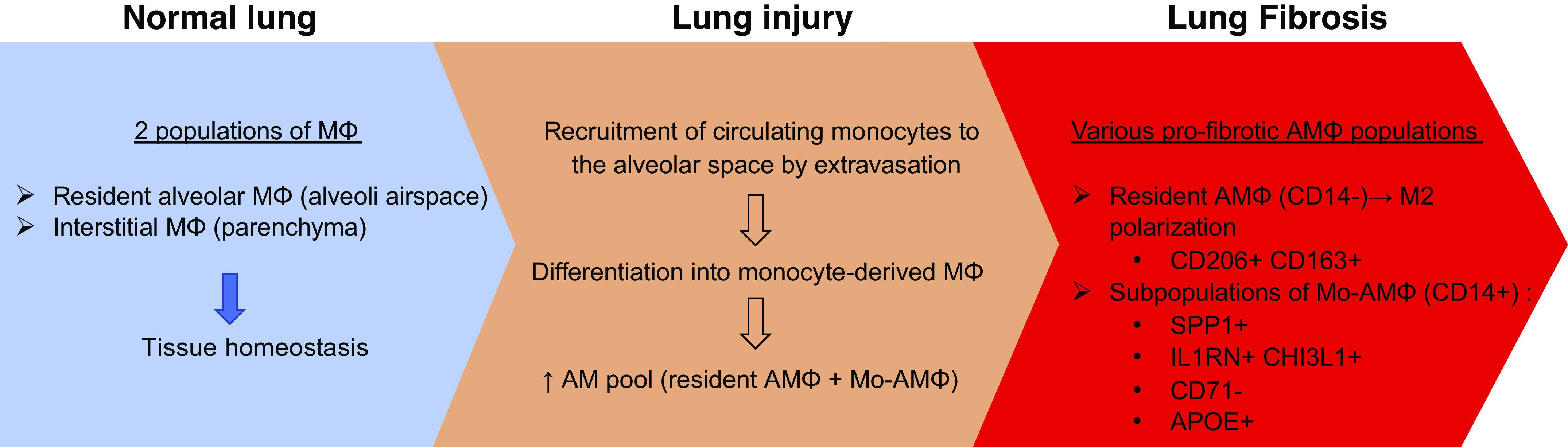
Pulmonary fibrosis results from a plethora of abnormal pathogenetic events. In idiopathic pulmonary fibrosis (IPF), inhalational, environmental, or occupational exposures in genetically and epigenetically predisposed individuals trigger recurrent cycles of alveolar epithelial cell injury, activation of coagulation pathways, chemoattraction, and differentiation of monocytes into monocyte-derived alveolar macrophages (Mo-AMs). When these events happen intermittently and repeatedly throughout the individual's life cycle, the wound repair process becomes aberrant leading to bronchiolization of distal air spaces, fibroblast accumulation, extracellular matrix deposition, and loss of the alveolar-capillary architecture. The role of immune dysregulation in IPF pathogenesis and progression has been underscored in the past mainly after the disappointing results of immunosuppressant use in IPF patients; however, recent reports highlighting the prognostic and mechanistic roles of monocytes and Mo-AMs revived the interest in immune dysregulation in IPF. In this review, we will discuss the role of these cells in the onset and progression of IPF, as well as potential targeted therapies.
肺纤维化是由大量异常的病理事件引起的。在特发性肺纤维化(IPF)中,遗传和表观遗传易感性个体暴露于吸入性、环境性或职业性因素,会引发反复的肺泡上皮细胞损伤、凝血途径激活、趋化作用和单核细胞分化为单核细胞源性肺泡巨噬细胞(Mo-AMs)。当这些事件在个体的生命周期中间歇性和反复发生时,伤口修复过程会变得异常,导致远端气腔的细支气管化、成纤维细胞积聚、细胞外基质沉积以及肺泡毛细血管结构的丧失。免疫失调在 IPF 发病机制和进展中的作用在过去主要是在 IPF 患者使用免疫抑制剂的结果令人失望之后得到强调的;然而,最近的报告强调了单核细胞和 Mo-AMs 的预后和机制作用,重新引起了人们对 IPF 中免疫失调的兴趣。在这篇综述中,我们将讨论这些细胞在 IPF 的发病和进展中的作用,以及潜在的靶向治疗方法。