Martin-Geary Alexandra C, Blakes Alexander J M, Dawes Ruebena, Findlay Scott D, Lord Jenny, Walker Susan, Talbot-Martin Jonathan, Wieder Nechama, D'Souza Elston N, Fernandes Maria, Hilton Sarah, Lahiri Nayana, Campbell Christopher, Jenkinson Sarah, DeGoede Christian G E L, Anderson Emily R, Burge Christopher B, Sanders Stephan J, Ellingford Jamie, Baralle Diana, Banka Siddharth, Whiffin Nicola
Big Data Institute, University of Oxford, UK.
Wellcome Centre for Human Genetics, University of Oxford, UK.
medRxiv. 2023 Sep 12:2023.09.12.23295416. doi: 10.1101/2023.09.12.23295416.
Both promoters and untranslated regions (UTRs) have critical regulatory roles, yet variants in these regions are largely excluded from clinical genetic testing due to difficulty in interpreting pathogenicity. The extent to which these regions may harbour diagnoses for individuals with rare disease is currently unknown.
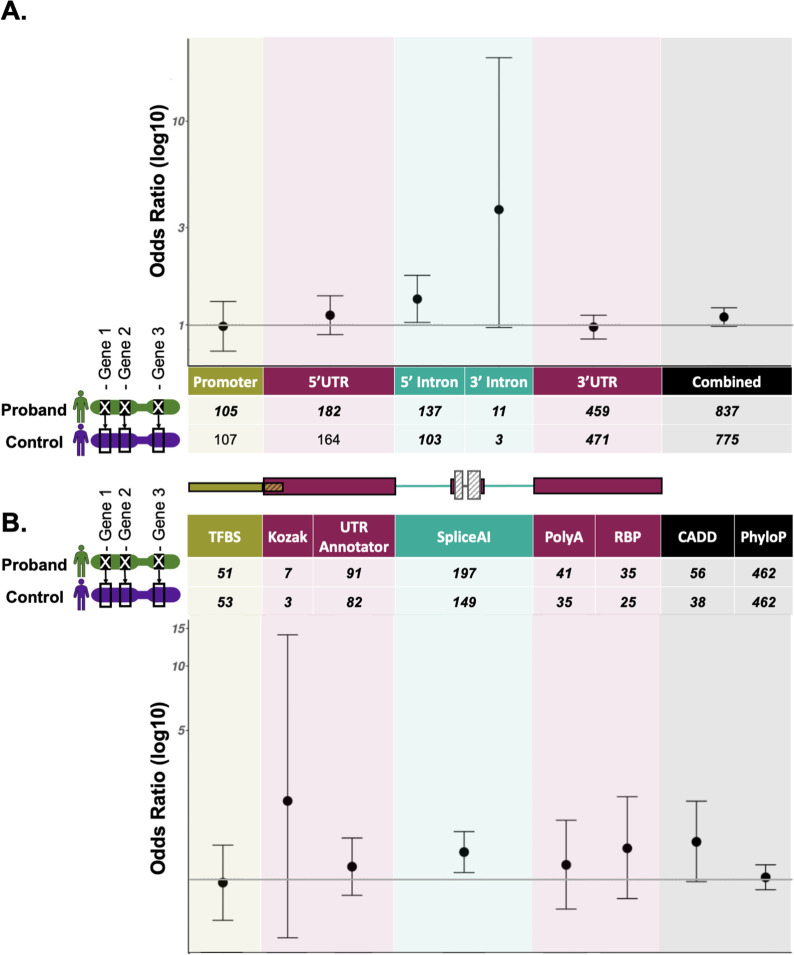
We present a framework for the identification and annotation of potentially deleterious proximal promoter and UTR variants in known dominant disease genes. We use this framework to annotate variants (DNVs) in 8,040 undiagnosed individuals in the Genomics England 100,000 genomes project, which were subject to strict region-based filtering, clinical review, and validation studies where possible. In addition, we performed region and variant annotation-based burden testing in 7,862 unrelated probands against matched unaffected controls.
We prioritised eleven DNVs and identified an additional variant overlapping one of the eleven. Ten of these twelve variants (82%) are in genes that are a strong match to the individual's phenotype and six had not previously been identified. Through burden testing, we did not observe a significant enrichment of potentially deleterious promoter and/or UTR variants in individuals with rare disease collectively across any of our region or variant annotations.
Overall, we demonstrate the value of screening promoters and UTRs to uncover additional diagnoses for previously undiagnosed individuals with rare disease and provide a framework for doing so without dramatically increasing interpretation burden.
启动子和非翻译区(UTR)均具有关键的调控作用,但由于难以解释致病性,这些区域的变异在很大程度上被排除在临床基因检测之外。目前尚不清楚这些区域可能为罕见病个体提供诊断的程度。
我们提出了一个框架,用于识别和注释已知显性疾病基因中潜在有害的近端启动子和UTR变异。我们使用这个框架对英国基因组学10万基因组计划中8040名未确诊个体的变异(DNV)进行注释,这些变异经过了严格的基于区域的筛选、临床评估,并在可能的情况下进行了验证研究。此外,我们在7862名无关先证者与匹配的未受影响对照中进行了基于区域和变异注释的负担测试。
我们对11个DNV进行了优先级排序,并确定了另外一个与这11个中的一个重叠的变异。这12个变异中有10个(82%)位于与个体表型高度匹配的基因中,其中6个此前未被发现。通过负担测试,在我们的任何区域或变异注释中,我们都没有观察到罕见病个体中潜在有害启动子和/或UTR变异的显著富集。
总体而言,我们证明了筛选启动子和UTR对于为先前未确诊的罕见病个体发现额外诊断的价值,并提供了一个在不显著增加解释负担的情况下进行此操作的框架。