Rubert Diego P, Braga Marília D V
Faculdade de Computação, Universidade Federal de Mato Grosso do Sul, Campo Grande, Brazil.
Faculty of Technology and Center for Biotechnology (CeBiTec), Bielefeld University, Bielefeld, Germany.
Algorithms Mol Biol. 2023 Sep 28;18(1):14. doi: 10.1186/s13015-023-00238-y.
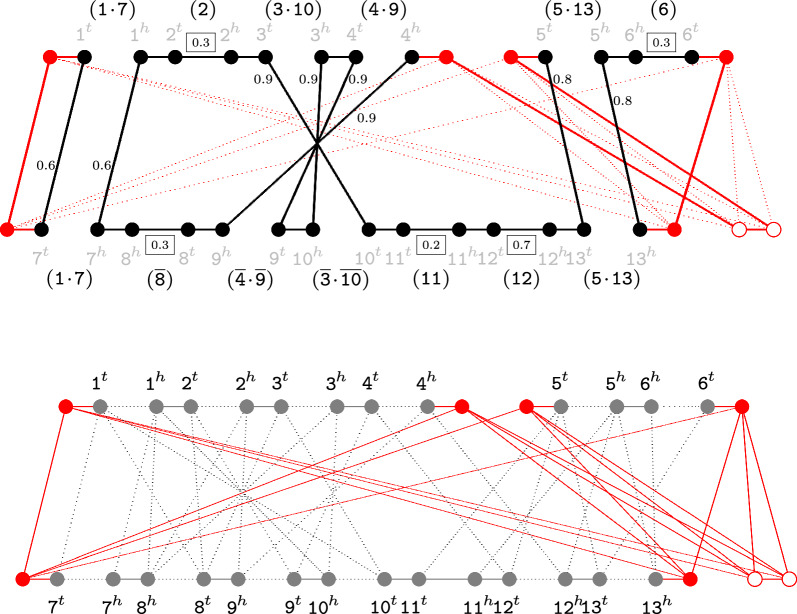
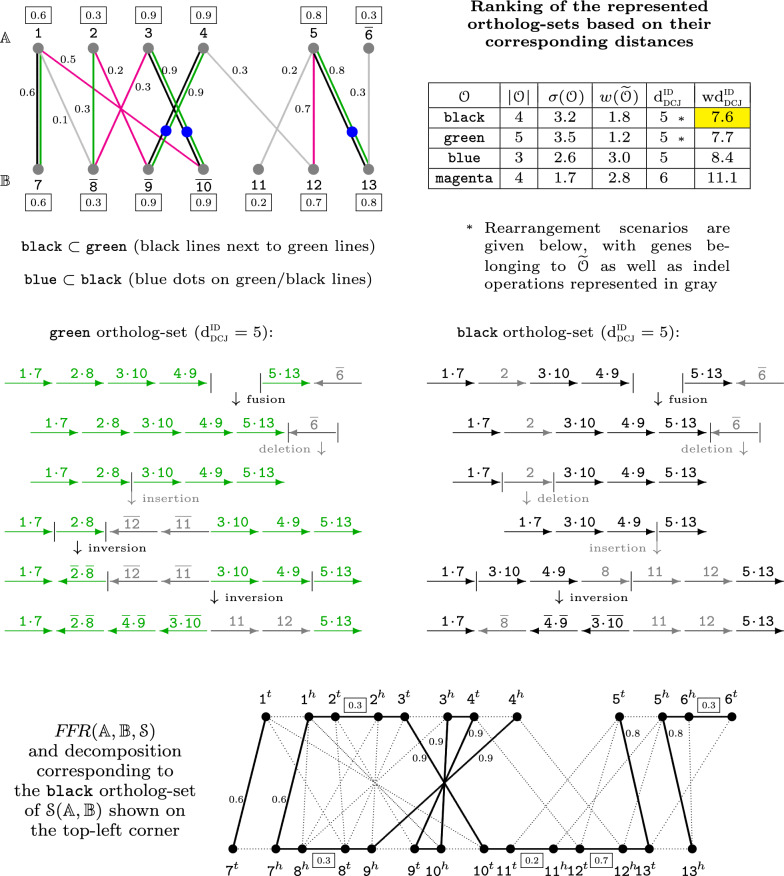
Recently we developed a gene orthology inference tool based on genome rearrangements (Journal of Bioinformatics and Computational Biology 19:6, 2021). Given a set of genomes our method first computes all pairwise gene similarities. Then it runs pairwise ILP comparisons to compute optimal gene matchings, which minimize, by taking the similarities into account, the weighted rearrangement distance between the analyzed genomes (a problem that is NP-hard). The gene matchings are then integrated into gene families in the final step. The mentioned ILP includes an optimal capping that connects each end of a linear segment of one genome to an end of a linear segment in the other genome, producing an exponential increase of the search space.
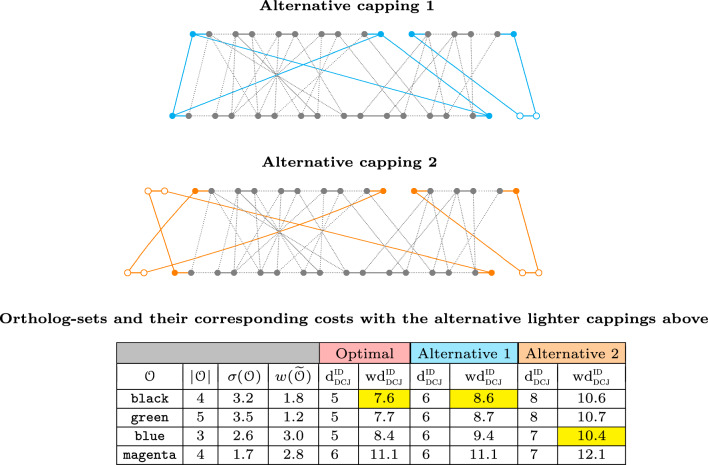
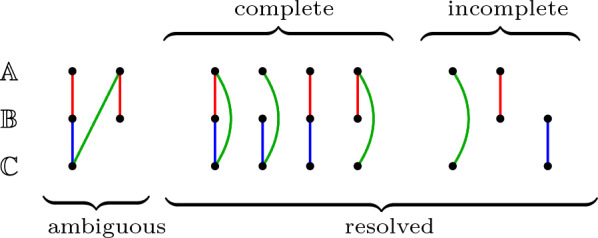
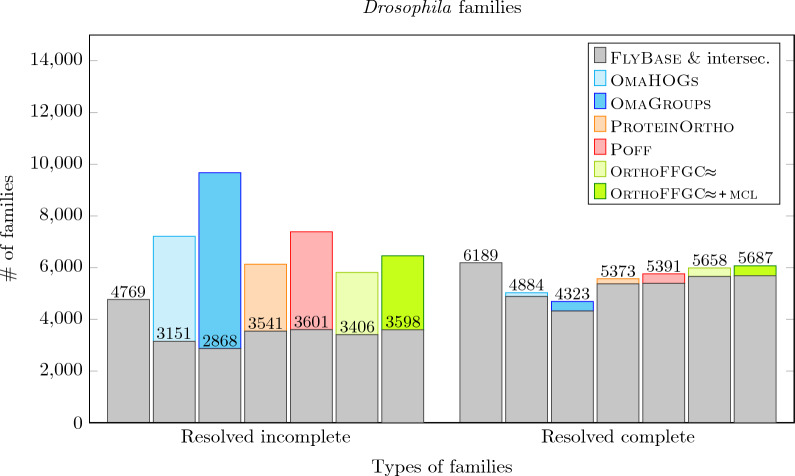
In this work, we design and implement a heuristic capping algorithm that replaces the optimal capping by clustering (based on their gene content intersections) the linear segments into [Formula: see text] subsets, whose ends are capped independently. Furthermore, in each subset, instead of allowing all possible connections, we let only the ends of content-related segments be connected. Although there is no guarantee that m is much bigger than one, and with the possible side effect of resulting in sub-optimal instead of optimal gene matchings, the heuristic works very well in practice, from both the speed performance and the quality of computed solutions. Our experiments on primate and fruit fly genomes show two positive results. First, for complete assemblies of five primates the version with heuristic capping reports orthologies that are very similar to the orthologies computed by the version of our tool with optimal capping. Second, we were able to efficiently analyze fruit fly genomes with incomplete assemblies distributed in hundreds or even thousands of contigs, obtaining gene families that are very similar to [Formula: see text] families. Indeed, our tool inferred a higher number of complete cliques, with a higher intersection with [Formula: see text], when compared to gene families computed by other inference tools. We added a post-processing for refining, with the aid of the [Formula: see text] algorithm, our ambiguous families (those with more than one gene per genome), improving even more the accuracy of our results. Our approach is implemented into a pipeline incorporating the pre-computation of gene similarities and the post-processing refinement of ambiguous families with [Formula: see text]. Both the original version with optimal capping and the new modified version with heuristic capping can be downloaded, together with their detailed documentations, at https://gitlab.ub.uni-bielefeld.de/gi/FFGC or as a Conda package at https://anaconda.org/bioconda/ffgc .
最近我们开发了一种基于基因组重排的基因直系同源推断工具(《生物信息学与计算生物学杂志》,2021年第19卷第6期)。给定一组基因组,我们的方法首先计算所有基因对之间的相似度。然后运行成对的整数线性规划(ILP)比较来计算最优基因匹配,通过考虑相似度来最小化分析基因组之间的加权重排距离(这是一个NP难问题)。最后一步将基因匹配整合到基因家族中。上述ILP包括一种最优封顶,即将一个基因组线性片段的两端连接到另一个基因组线性片段的一端,这使得搜索空间呈指数级增长。
在这项工作中,我们设计并实现了一种启发式封顶算法,该算法通过将线性片段聚类(基于它们的基因内容交集)为[公式:见原文]个子集来取代最优封顶,这些子集的末端独立封顶。此外,在每个子集中,我们不是允许所有可能的连接,而是只连接内容相关片段的末端。尽管不能保证m比1大很多,并且可能会导致产生次优而非最优的基因匹配,但从速度性能和计算解的质量来看,这种启发式方法在实践中效果很好。我们在灵长类动物和果蝇基因组上的实验显示了两个积极的结果。第一,对于五种灵长类动物的完整组装,采用启发式封顶的版本报告的直系同源关系与我们工具采用最优封顶版本计算的直系同源关系非常相似。第二,我们能够有效地分析分布在数百甚至数千个重叠群中的不完整组装的果蝇基因组,获得与[公式:见原文]家族非常相似的基因家族。事实上,与其他推断工具计算的基因家族相比,我们的工具推断出了更多的完整团簇,与[公式:见原文]的交集更高。我们借助[公式:见原文]算法对模糊家族(每个基因组有多个基因的家族)进行后处理以优化,进一步提高了结果的准确性。我们的方法被实现为一个管道,其中包括基因相似度的预计算以及使用[公式:见原文]对模糊家族进行后处理优化。带有最优封顶的原始版本和带有启发式封顶的新修改版本,连同它们的详细文档,都可以在https://gitlab.ub.uni-bielefeld.de/gi/FFGC上下载,或者作为一个Conda包在https://anaconda.org/bioconda/ffgc上下载。