Bohnenkämper Leonard, Stoye Jens, Doerr Daniel
Faculty of Technology, Bielefeld University, Universitätsstraße 25, 33615, Bielefeld, NRW, Germany.
Center for Biotechnology (CeBiTec), Bielefeld University, Universitätsstraße 25, 33615, Bielefeld, NRW, Germany.
Algorithms Mol Biol. 2025 Jun 7;20(1):10. doi: 10.1186/s13015-025-00279-5.
We study the classical problem of inferring ancestral genomes from a set of extant genomes under a given phylogeny, known as the Small Parsimony Problem (SPP). Genomes are represented as sequences of oriented markers, organized in one or more linear or circular chromosomes. Any marker may appear in several copies, without restriction on orientation or genomic location, known as the natural genomes model. Evolutionary events along the branches of the phylogeny encompass large scale rearrangements, including segmental inversions, translocations, gain and loss (DCJ-indel model). Even under simpler rearrangement models, such as the classical breakpoint model without duplicates, the SPP is computationally intractable. Nevertheless, the SPP for natural genomes under the DCJ-indel model has been studied recently, with limited success.
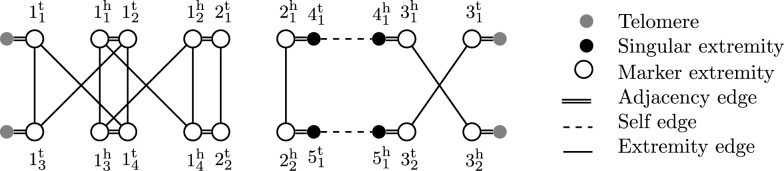
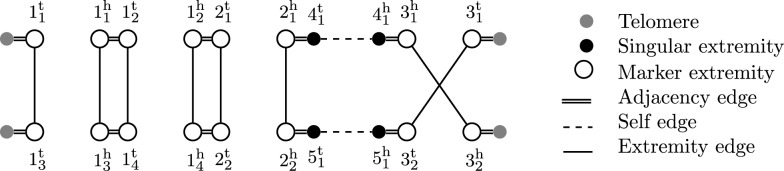
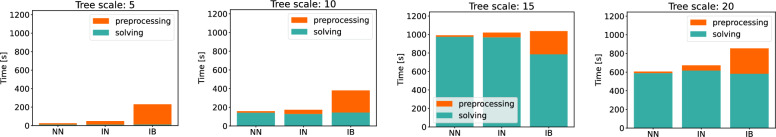
Building on prior work, we present a highly optimized ILP that is able to solve the SPP for sufficiently small phylogenies and gene families. A notable improvement w.r.t. the previous result is an optimized way of handling both circular and linear chromosomes. This is especially relevant to the SPP, since the chromosomal structure of ancestral genomes is unknown and the solution space for this chromosomal structure is typically large.
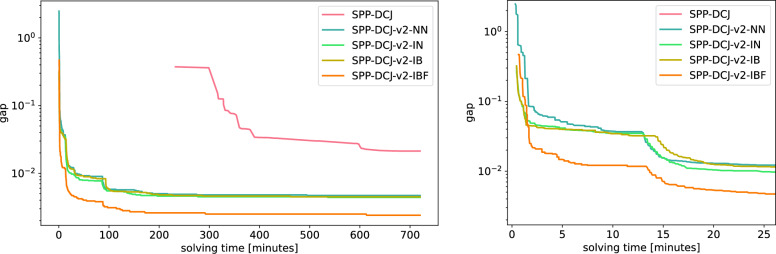
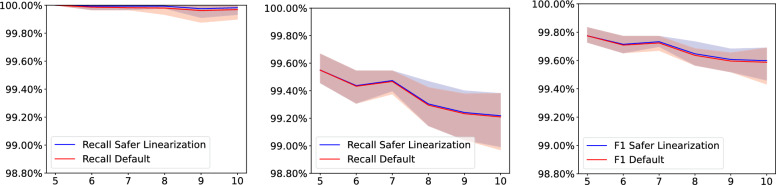
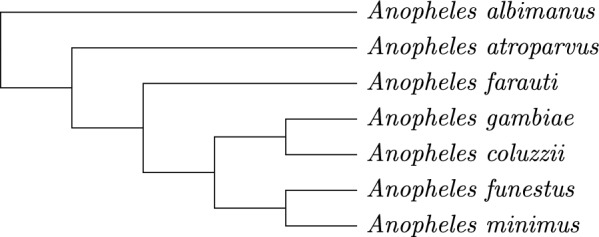
We benchmark our method on simulated and real data. On simulated phylogenies we observe a considerable performance improvement on problems that include linear chromosomes. And even when the ground truth contains only one circular chromosome per genome, our method outperforms its predecessor due to its optimized handling of the solution space. The practical advantage becomes also visible in an analysis of seven Anopheles taxa.
我们研究在给定系统发育树的情况下,从一组现存基因组推断祖先基因组的经典问题,即所谓的小简约问题(SPP)。基因组被表示为定向标记的序列,组织在一条或多条线性或环状染色体中。任何标记可能会出现多个拷贝,对其方向或基因组位置没有限制,这就是自然基因组模型。沿着系统发育树分支的进化事件包括大规模重排,包括片段倒位、易位、获得和丢失(DCJ - 插入缺失模型)。即使在更简单的重排模型下,例如没有重复的经典断点模型,SPP在计算上也是难以处理的。然而,最近对DCJ - 插入缺失模型下自然基因组的SPP进行了研究,但取得的成功有限。
在先前工作的基础上,我们提出了一种高度优化的整数线性规划(ILP),它能够解决足够小的系统发育树和基因家族的SPP。相对于先前结果的一个显著改进是处理环状和线性染色体的优化方法。这与SPP特别相关,因为祖先基因组的染色体结构是未知的,并且这种染色体结构的解空间通常很大。
我们在模拟数据和真实数据上对我们的方法进行了基准测试。在模拟系统发育树上,我们观察到在包含线性染色体的问题上性能有相当大的提升。而且即使在每个基因组的真实情况仅包含一条环状染色体的情况下,由于我们的方法对解空间的优化处理,它也优于其前身。在对七个按蚊类群的分析中,实际优势也很明显。