Xingtai People's Hospital Affiliated to Hebei Medical University, Xingtai City, Hebei Province, China.
Ningbo Huamei Hospital, University of Chinese Academy of Sciences, Ningbo City, Zhejiang Province, China.
BMC Med Genomics. 2023 Oct 7;16(1):236. doi: 10.1186/s12920-023-01672-y.
Osteoarthritis (OA) is a multifaceted chronic joint disease characterized by complex mechanisms. It has a detrimental impact on the quality of life for individuals in the middle-aged and elderly population while also imposing a significant socioeconomic burden. At present, there remains a lack of comprehensive understanding regarding the pathophysiology of OA. The objective of this study was to examine the genes, functional pathways, and immune infiltration characteristics associated with the development and advancement of OA.
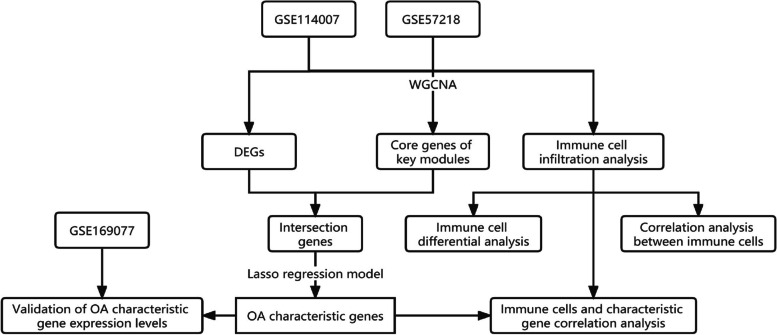
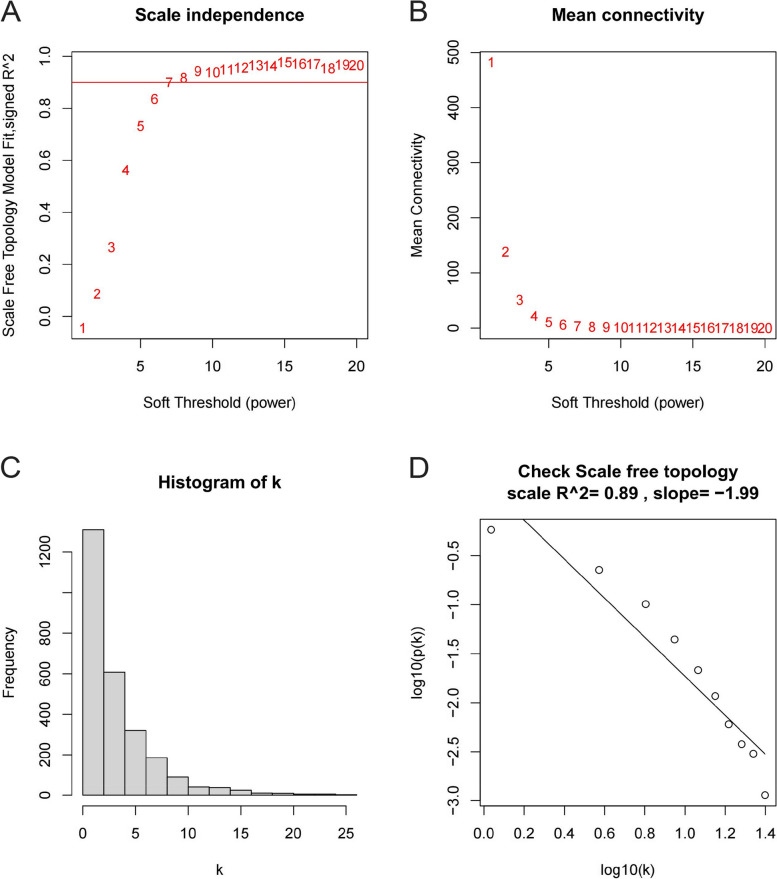
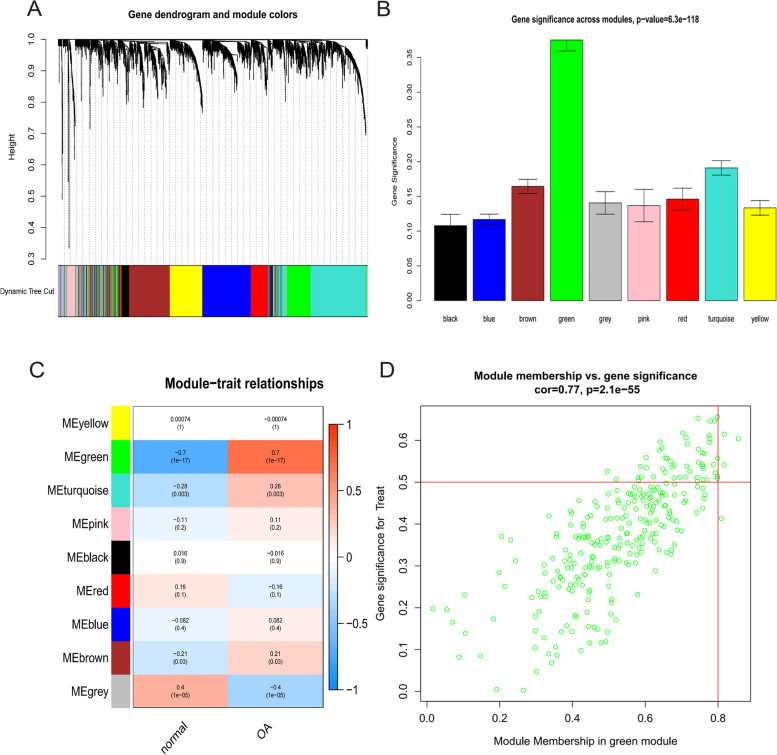
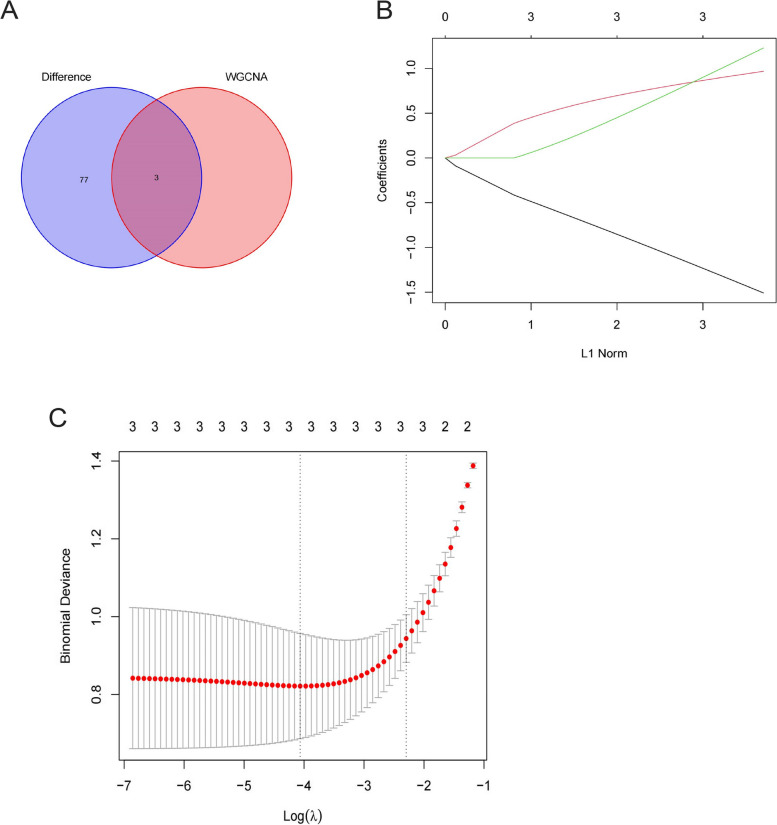
The Gene Expression Omnibus (GEO) database was utilized to acquire gene expression profiles. The R software was employed to conduct the screening of differentially expressed genes (DEGs) and perform enrichment analysis on these genes. The OA-characteristic genes were identified using the Weighted Gene Co-expression Network Analysis (WGCNA) and the Lasso algorithm. In addition, the infiltration levels of immune cells in cartilage were assessed using single-sample gene set enrichment analysis (ssGSEA). Subsequently, a correlation analysis was conducted to examine the relationship between immune cells and the OA-characteristic genes.
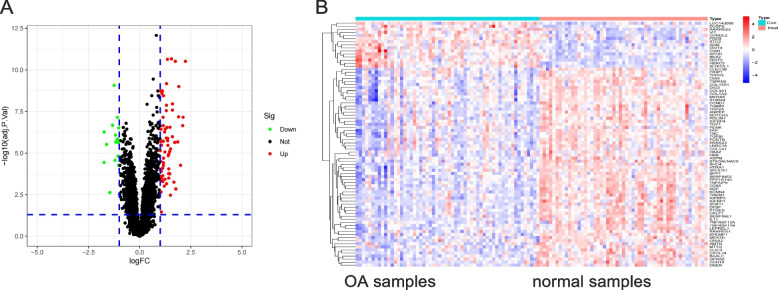
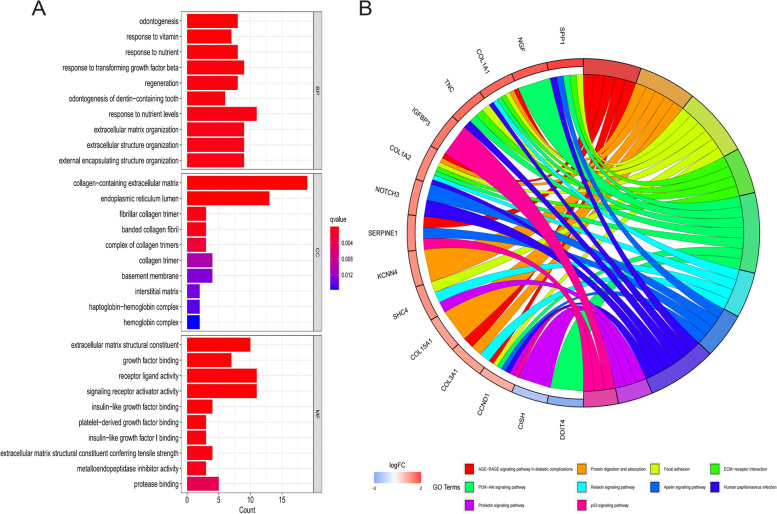
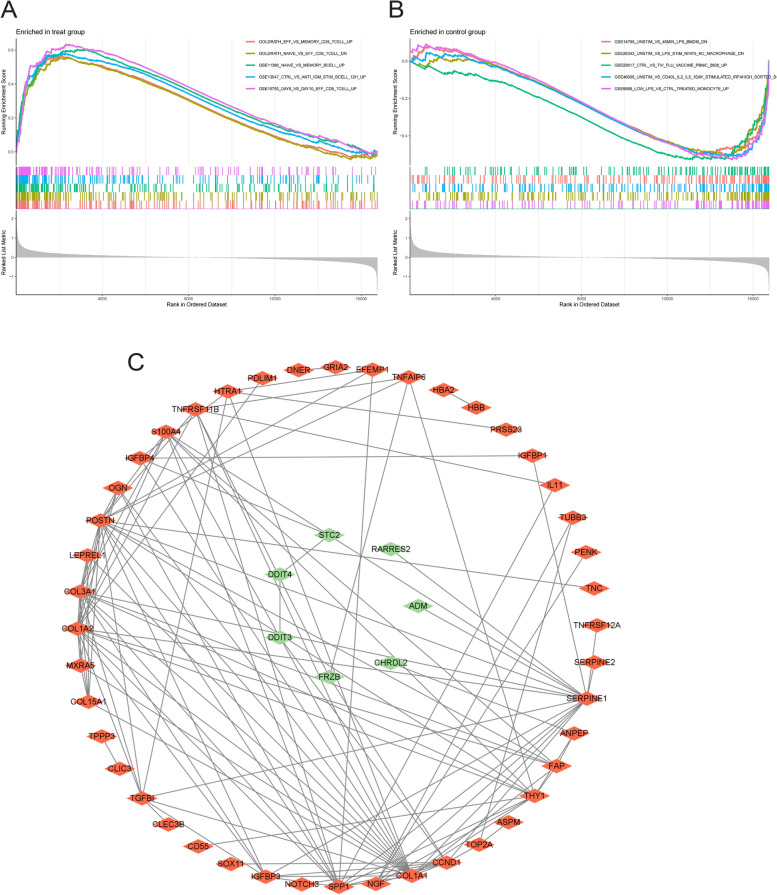
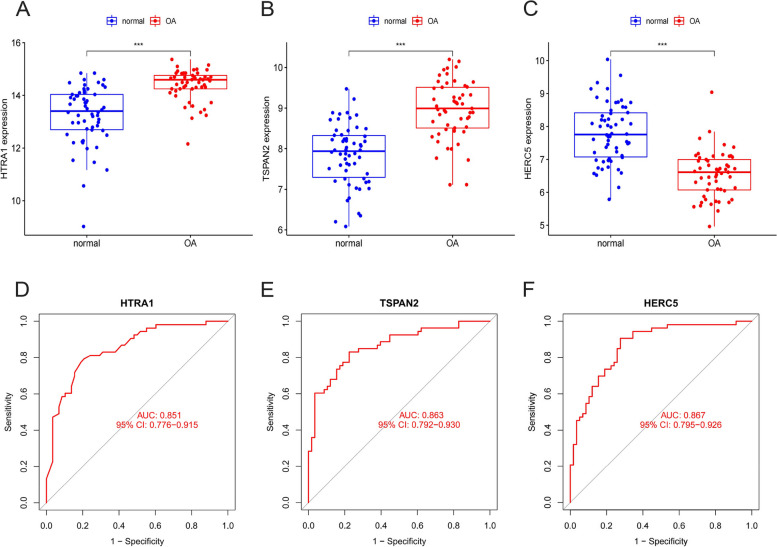
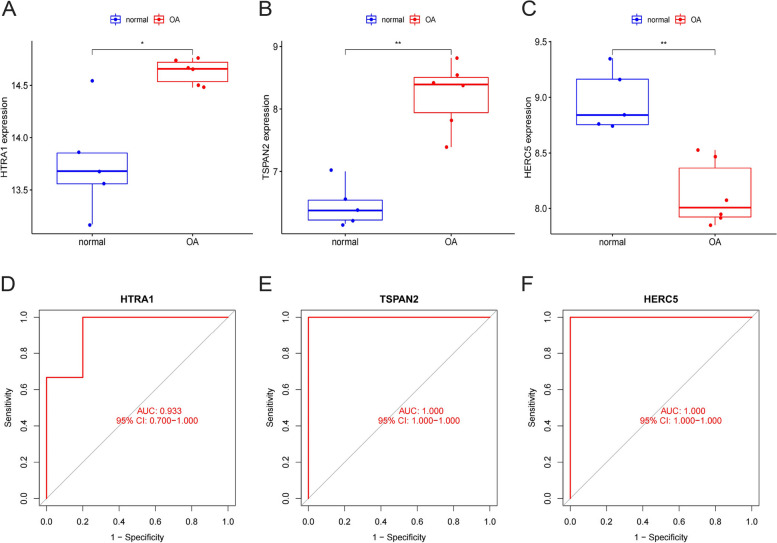
A total of 80 DEGs were identified. As determined by functional enrichment, these DEGs were associated with chondrocyte metabolism, apoptosis, and inflammation. Three OA-characteristic genes were identified using WGCNA and the lasso algorithm, and their expression levels were then validated using the verification set. Finally, the analysis of immune cell infiltration revealed that T cells and B cells were primarily associated with OA. In addition, Tspan2, HtrA1 demonstrated a correlation with some of the infiltrating immune cells.
The findings of an extensive bioinformatics analysis revealed that OA is correlated with a variety of distinct genes, functional pathways, and processes involving immune cell infiltration. The present study has successfully identified characteristic genes and functional pathways that hold potential as biomarkers for guiding drug treatment and facilitating molecular-level research on OA.
骨关节炎(OA)是一种多方面的慢性关节疾病,其特征是复杂的机制。它对中年和老年人群的生活质量有不利影响,同时也给社会经济带来了重大负担。目前,人们对 OA 的病理生理学仍缺乏全面的了解。本研究旨在探讨与 OA 发展和进展相关的基因、功能途径和免疫浸润特征。
使用基因表达综合(GEO)数据库获取基因表达谱。使用 R 软件筛选差异表达基因(DEGs)并对这些基因进行富集分析。使用加权基因共表达网络分析(WGCNA)和套索算法识别 OA 特征基因。此外,使用单样本基因集富集分析(ssGSEA)评估软骨中免疫细胞的浸润水平。随后,进行相关性分析以研究免疫细胞与 OA 特征基因之间的关系。
共鉴定出 80 个 DEGs。通过功能富集分析,这些 DEGs与软骨细胞代谢、凋亡和炎症有关。使用 WGCNA 和套索算法识别出三个 OA 特征基因,并使用验证集验证其表达水平。最后,免疫细胞浸润分析表明 T 细胞和 B 细胞主要与 OA 相关。此外,Tspan2、HtrA1 与一些浸润免疫细胞表现出相关性。
广泛的生物信息学分析发现,OA 与多种不同的基因、功能途径和涉及免疫细胞浸润的过程相关。本研究成功鉴定了特征基因和功能途径,它们可能作为指导药物治疗和促进 OA 分子水平研究的生物标志物。