F. M. Kirby Neurobiology Center, Department of Neurology, Boston Children's Hospital, Harvard Medical School, Boston, MA, US.
Center for Neurobehavioral Genetics, Jane and Terry Semel Institute for Neuroscience and Human Behavior, Department of Psychiatry and Biobehavioral Sciences, David Geffen School of Medicine at University of California, Los Angeles, CA, USA.
Nat Med. 2023 Nov;29(11):2866-2884. doi: 10.1038/s41591-023-02566-3. Epub 2023 Oct 9.
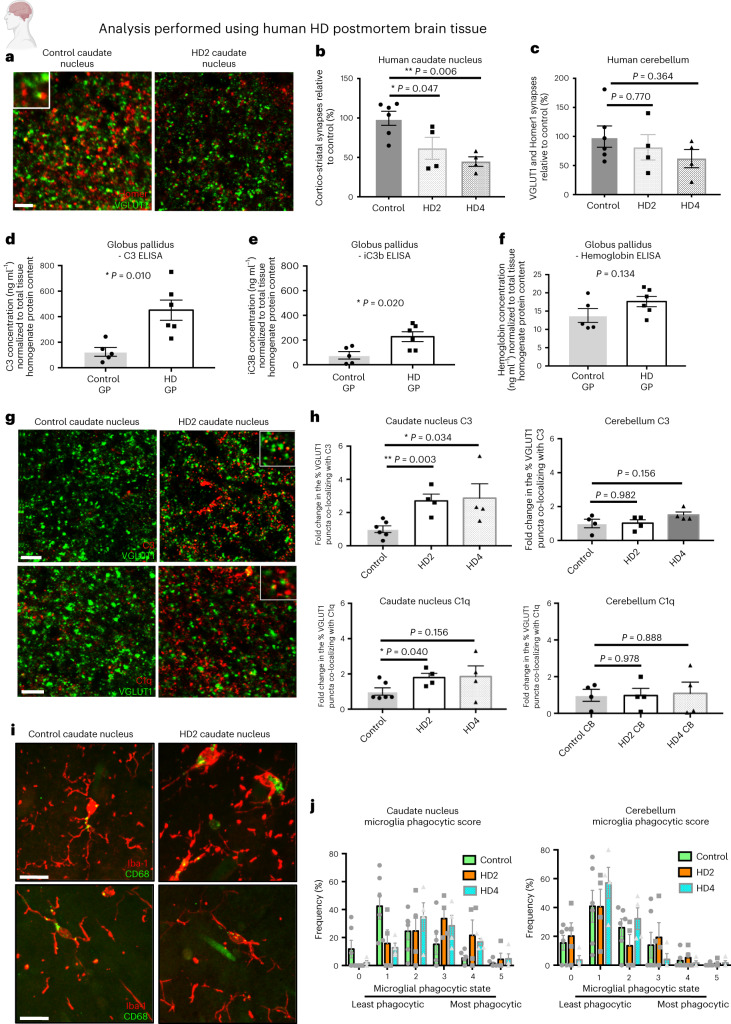
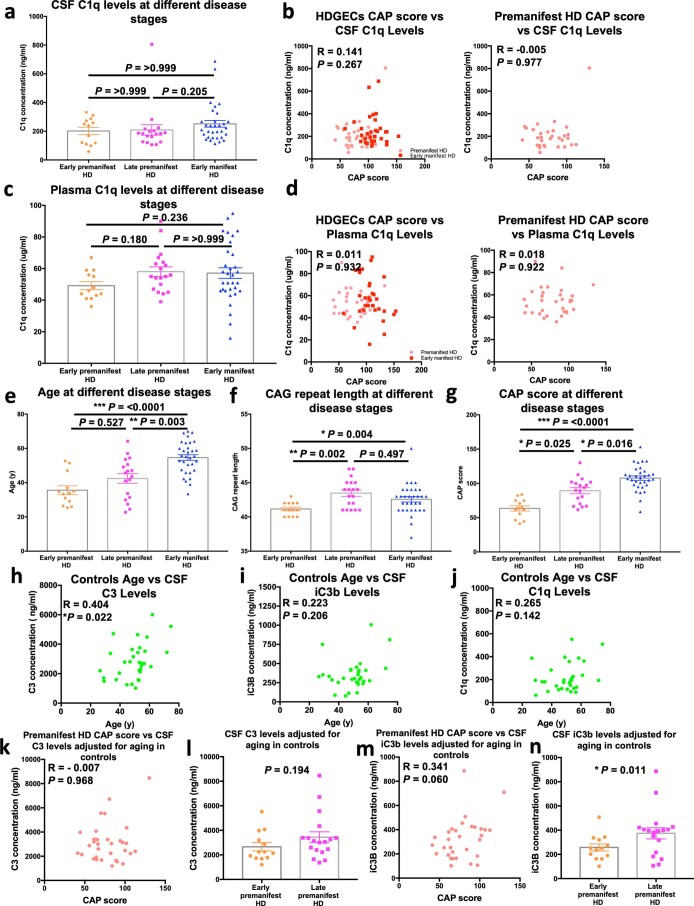
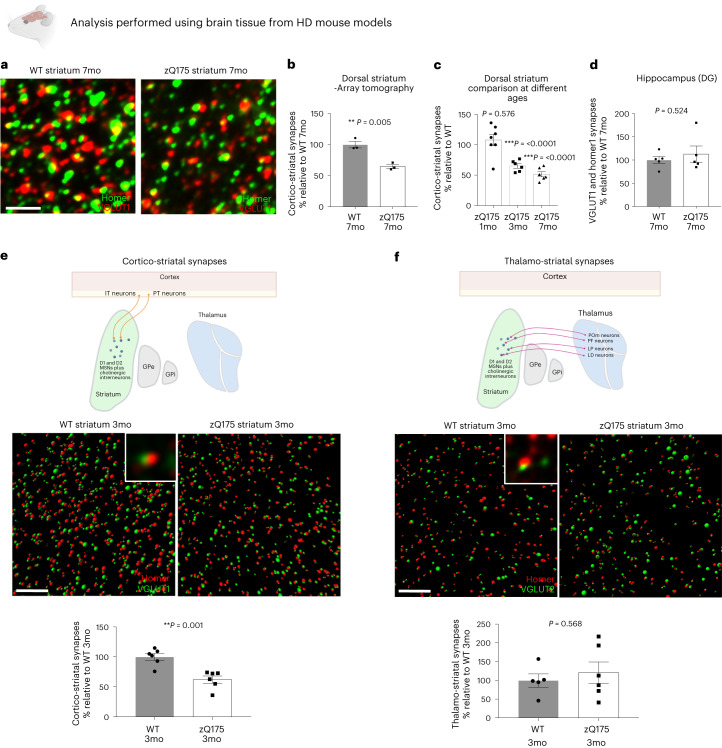
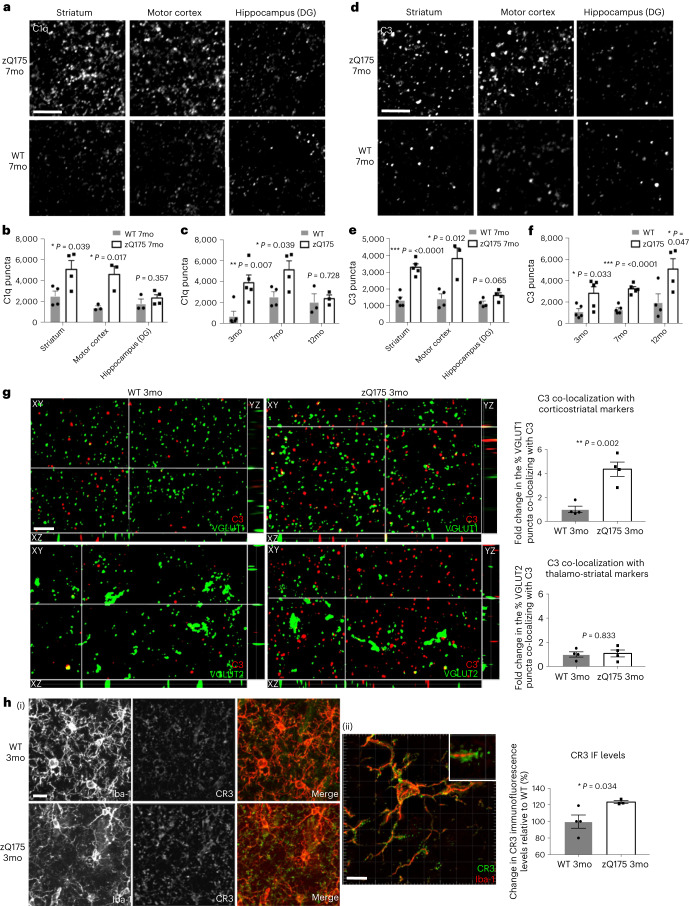
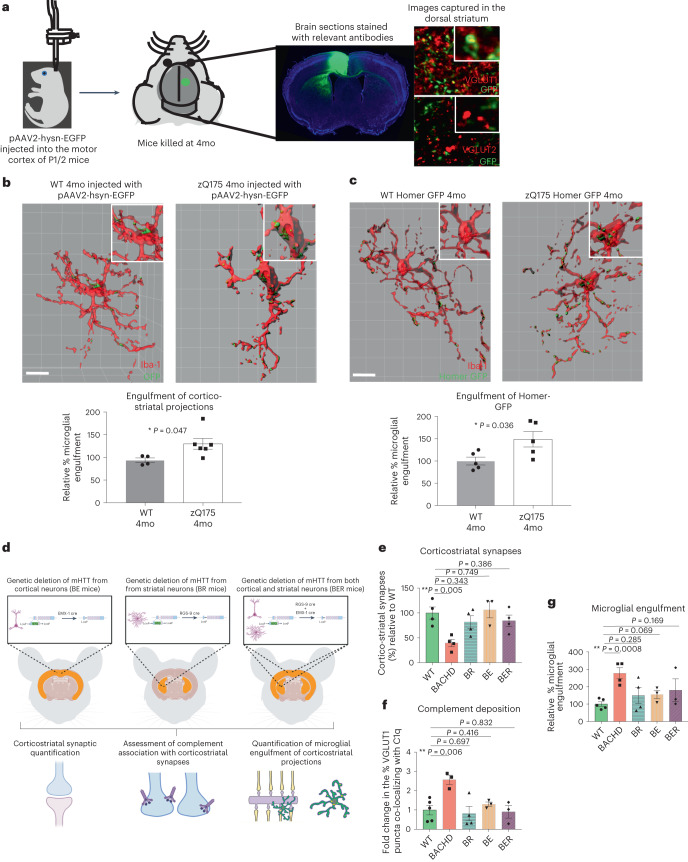
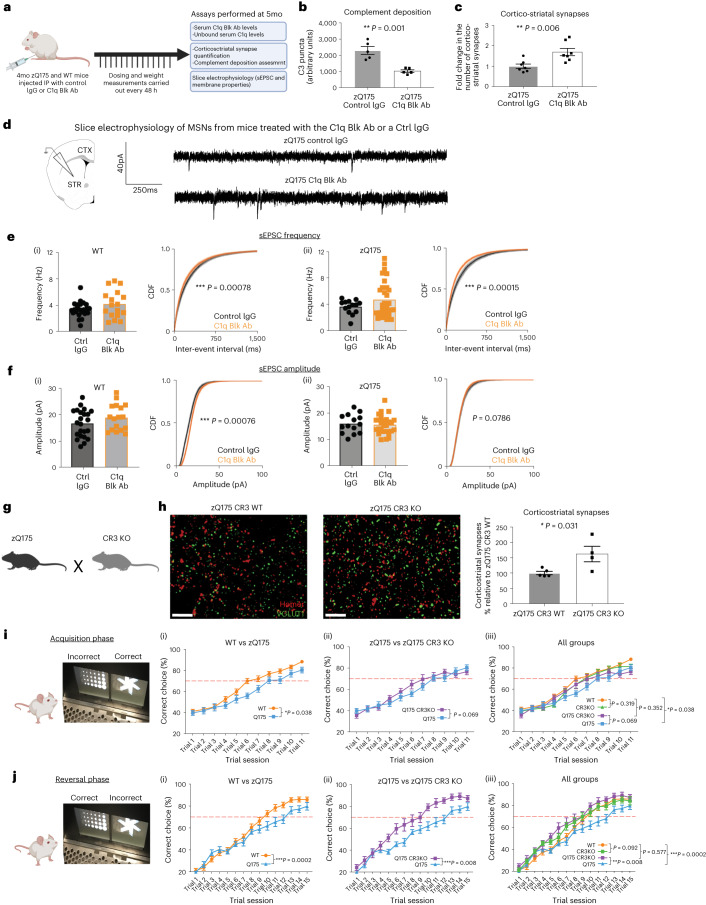
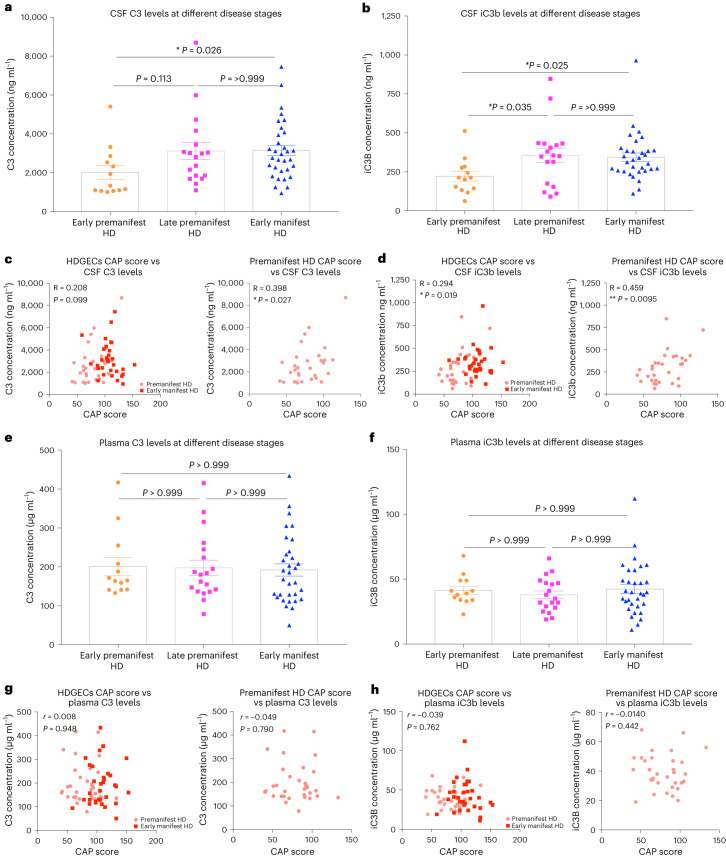
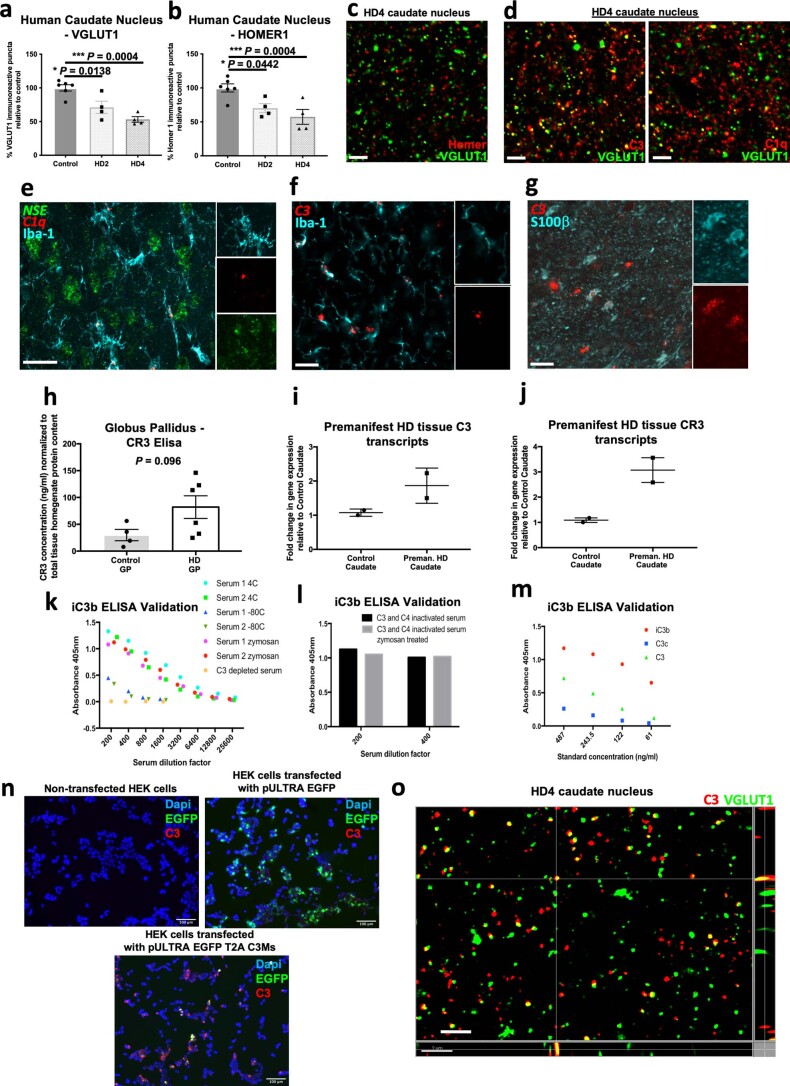
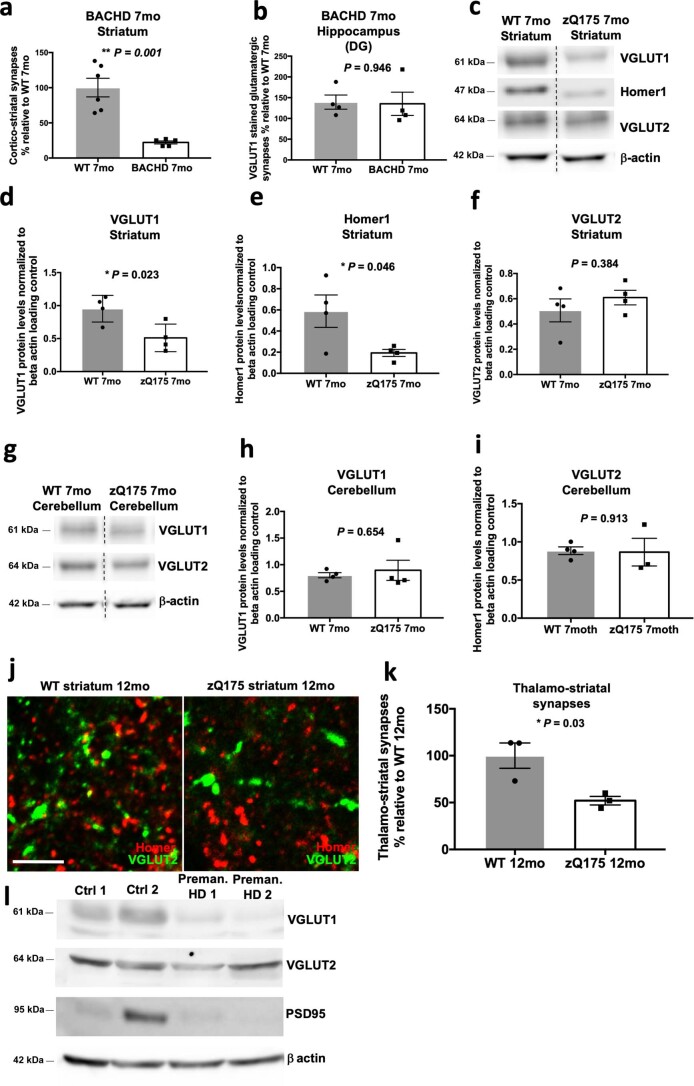
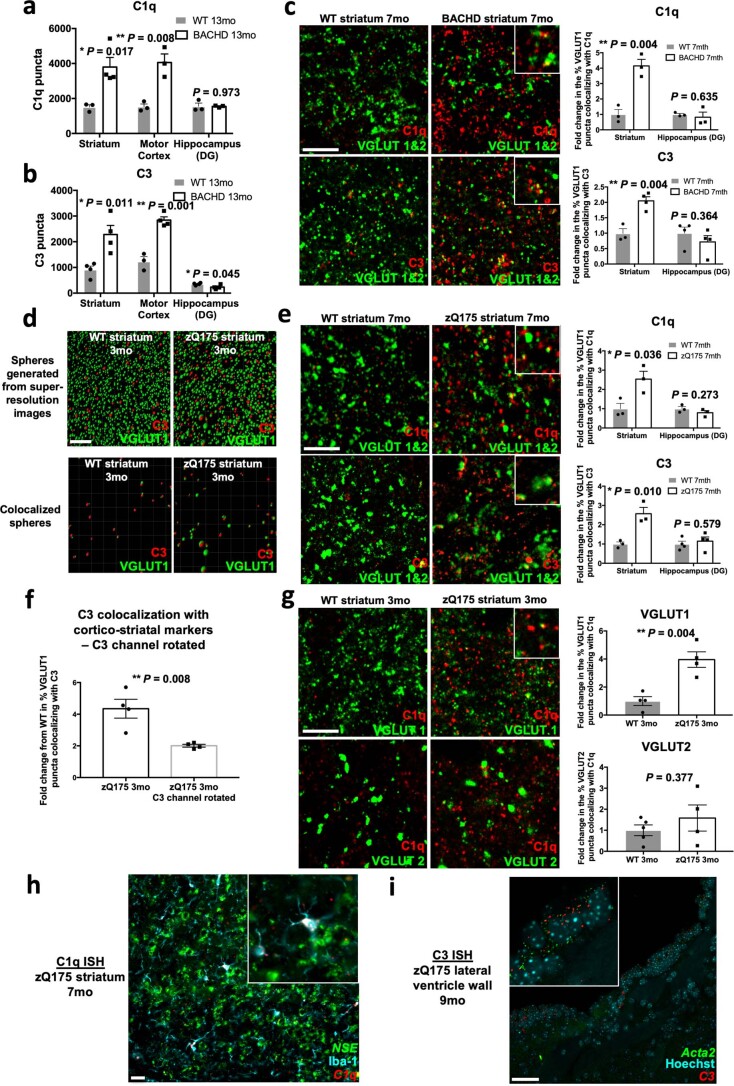
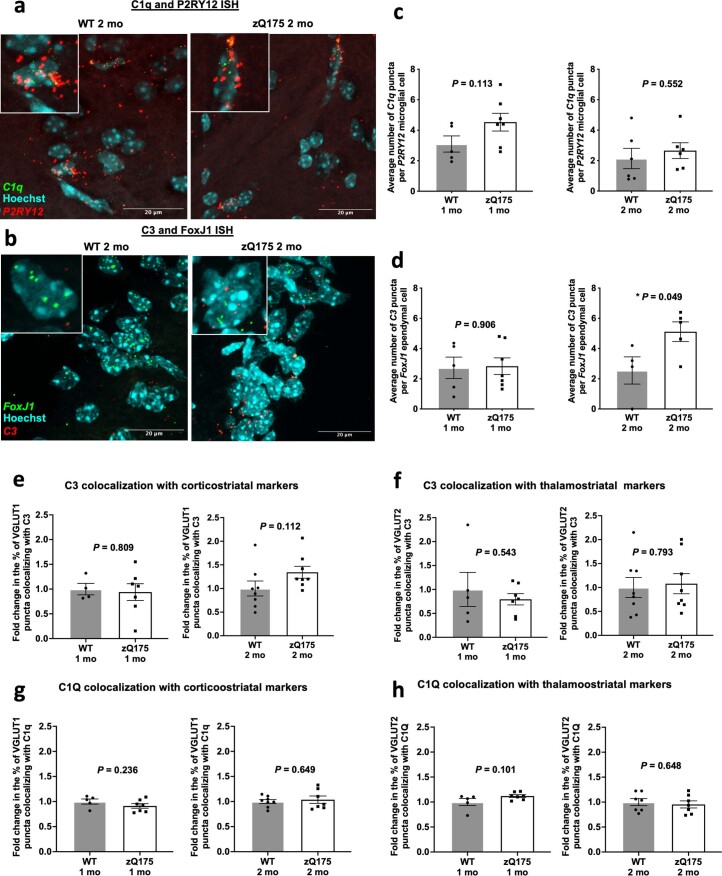
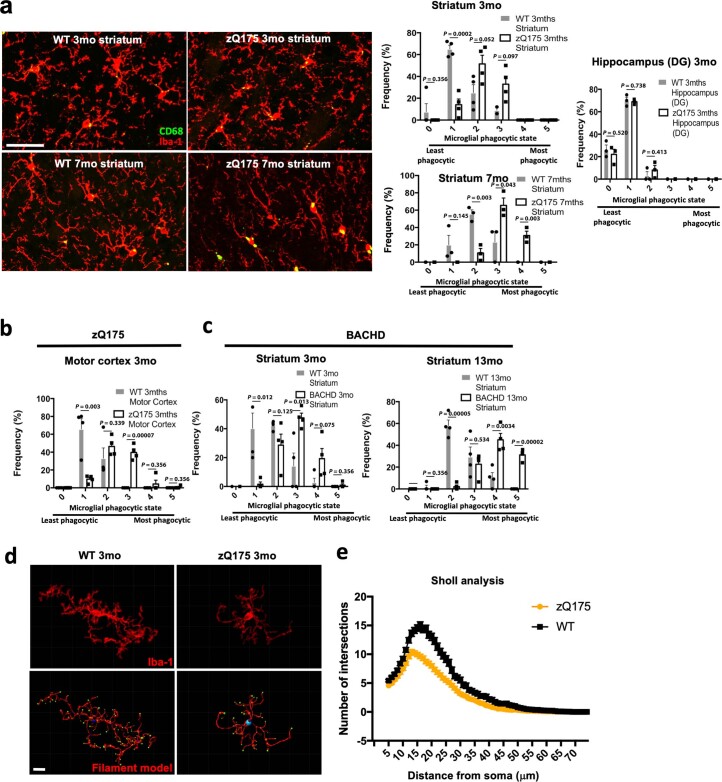
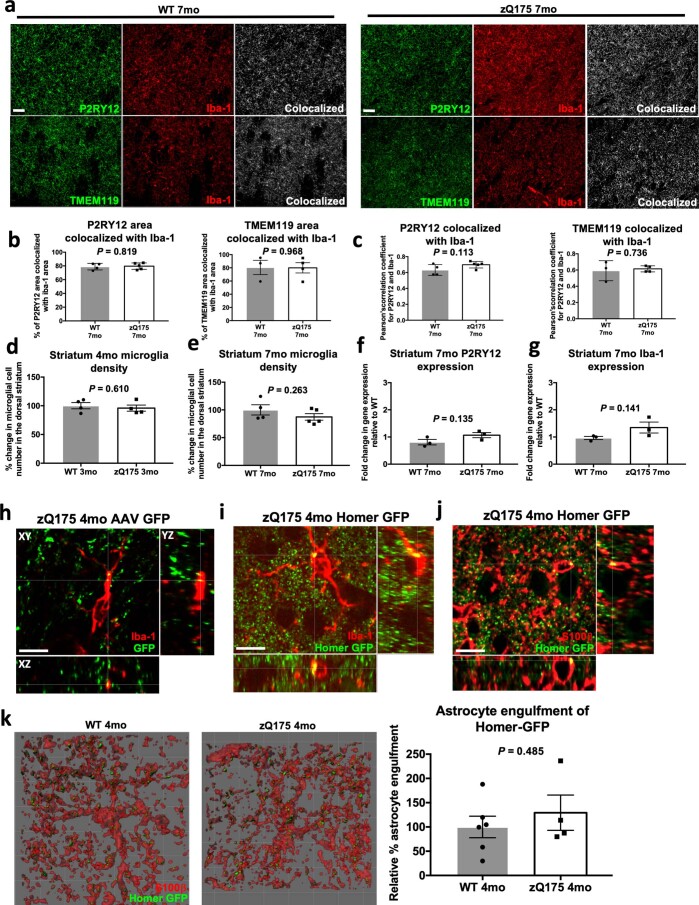
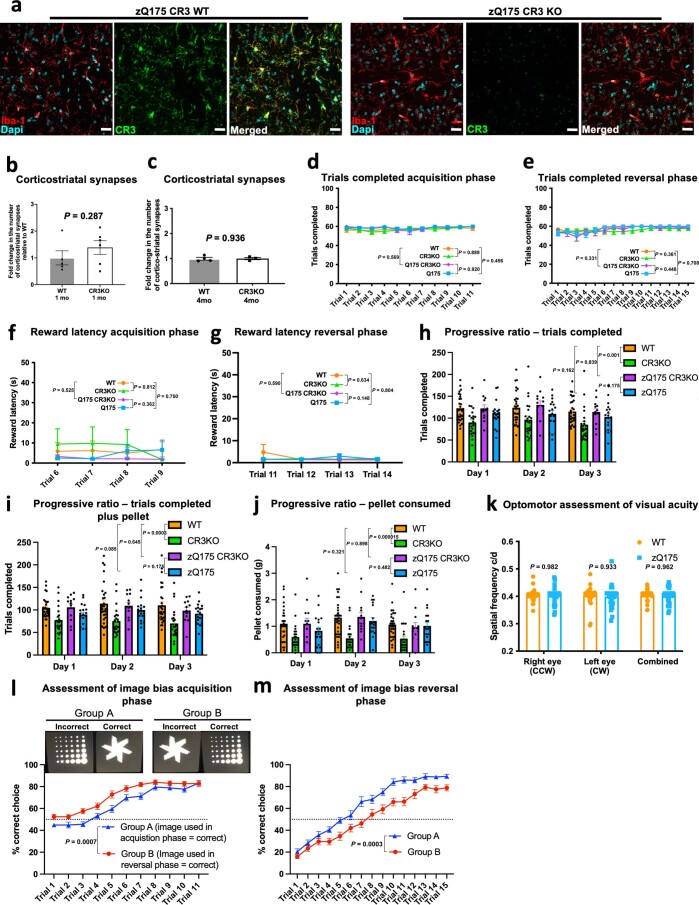
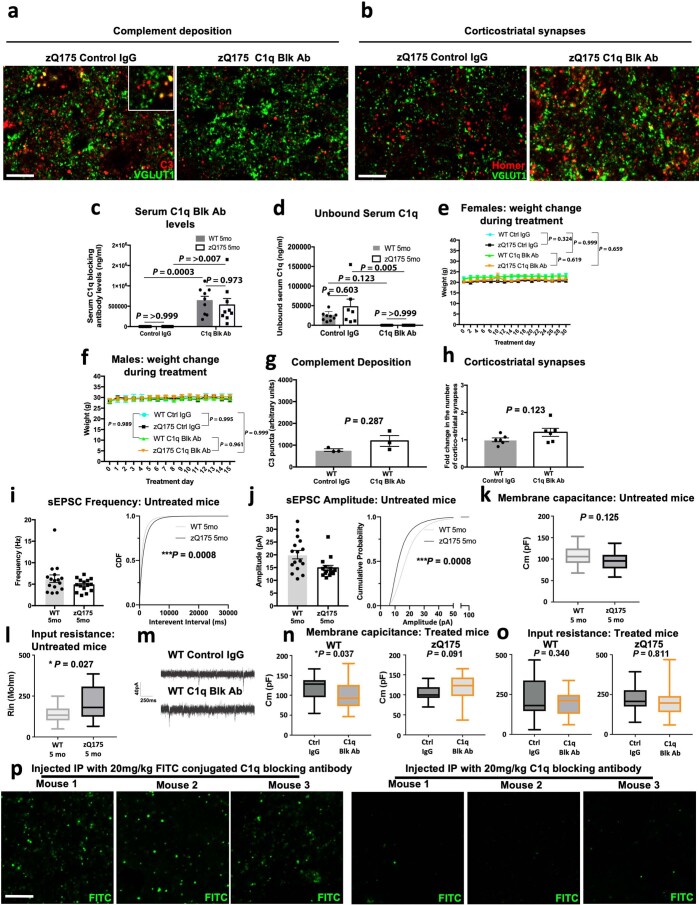
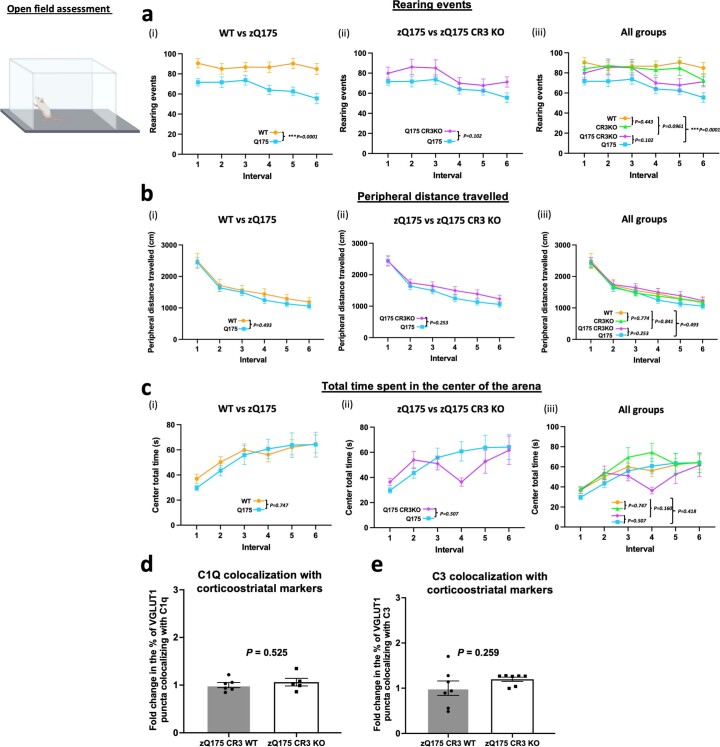
Huntington's disease (HD) is a devastating monogenic neurodegenerative disease characterized by early, selective pathology in the basal ganglia despite the ubiquitous expression of mutant huntingtin. The molecular mechanisms underlying this region-specific neuronal degeneration and how these relate to the development of early cognitive phenotypes are poorly understood. Here we show that there is selective loss of synaptic connections between the cortex and striatum in postmortem tissue from patients with HD that is associated with the increased activation and localization of complement proteins, innate immune molecules, to these synaptic elements. We also found that levels of these secreted innate immune molecules are elevated in the cerebrospinal fluid of premanifest HD patients and correlate with established measures of disease burden.In preclinical genetic models of HD, we show that complement proteins mediate the selective elimination of corticostriatal synapses at an early stage in disease pathogenesis, marking them for removal by microglia, the brain's resident macrophage population. This process requires mutant huntingtin to be expressed in both cortical and striatal neurons. Inhibition of this complement-dependent elimination mechanism through administration of a therapeutically relevant C1q function-blocking antibody or genetic ablation of a complement receptor on microglia prevented synapse loss, increased excitatory input to the striatum and rescued the early development of visual discrimination learning and cognitive flexibility deficits in these models. Together, our findings implicate microglia and the complement cascade in the selective, early degeneration of corticostriatal synapses and the development of cognitive deficits in presymptomatic HD; they also provide new preclinical data to support complement as a therapeutic target for early intervention.
亨廷顿病(HD)是一种破坏性的单基因神经退行性疾病,其特征是基底神经节中存在早期、选择性的病理学,尽管突变亨廷顿蛋白广泛表达。导致这种区域特异性神经元变性的分子机制以及这些机制如何与早期认知表型的发展相关,目前仍知之甚少。在这里,我们显示在 HD 患者的死后组织中,皮质和纹状体之间存在突触连接的选择性丧失,这与补体蛋白、先天免疫分子的激活和定位有关这些突触元件。我们还发现,这些分泌型先天免疫分子的水平在无症状 HD 患者的脑脊液中升高,并与疾病负担的既定测量指标相关。在 HD 的临床前遗传模型中,我们表明补体蛋白介导皮质纹状体突触在疾病发病早期的选择性消除,使它们被大脑常驻巨噬细胞群体的小胶质细胞清除。这个过程需要突变亨廷顿蛋白在皮质和纹状体神经元中表达。通过给予具有治疗相关性的 C1q 功能阻断抗体或遗传消融小胶质细胞上的补体受体来抑制这种补体依赖性消除机制,可以防止突触丢失,增加对纹状体的兴奋性输入,并挽救这些模型中视觉辨别学习和认知灵活性缺陷的早期发展。总之,我们的发现表明小胶质细胞和补体级联反应参与了皮质纹状体突触的选择性、早期退化以及在无症状 HD 中认知缺陷的发展;它们还提供了新的临床前数据,支持补体作为早期干预的治疗靶点。