Carsch Kurtis M, North Sasha C, DiMucci Ida M, Iliescu Andrei, Vojáčková Petra, Khazanov Thomas, Zheng Shao-Liang, Cundari Thomas R, Lancaster Kyle M, Betley Theodore A
Department of Chemistry and Chemical Biology, Harvard University Cambridge MA 02138 USA
Center for Advanced Scientific Computing and Modeling (CASCaM), Department of Chemistry, University of North Texas, Denton TX 76203 USA.
Chem Sci. 2023 Sep 26;14(39):10847-10860. doi: 10.1039/d3sc03641c. eCollection 2023 Oct 11.
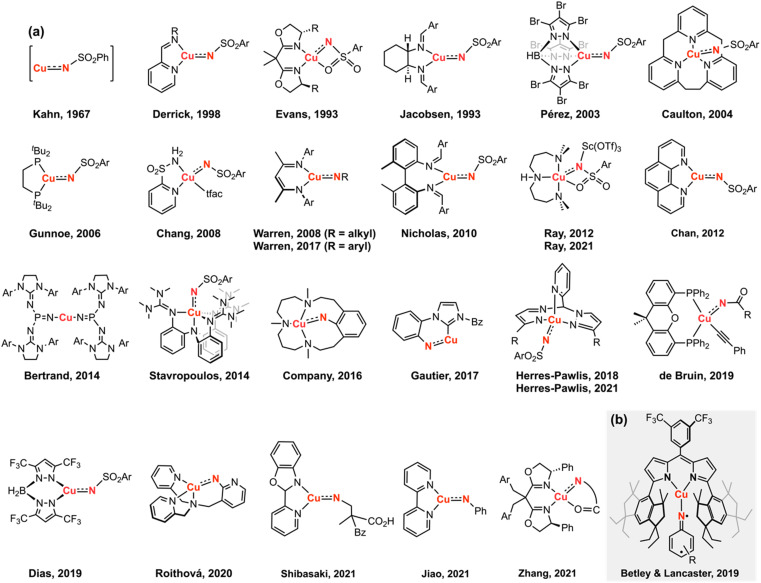
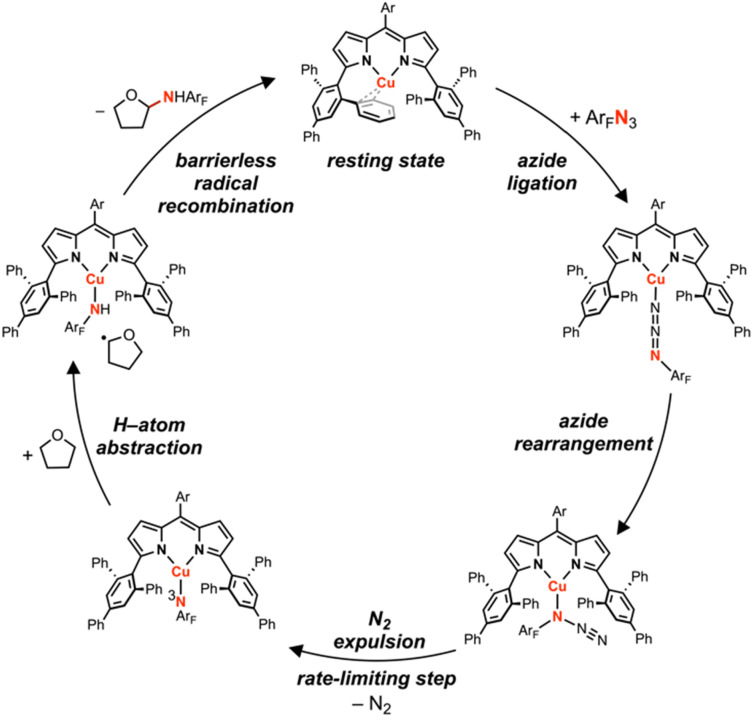
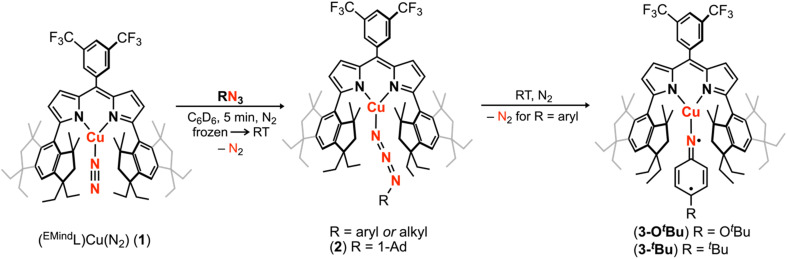
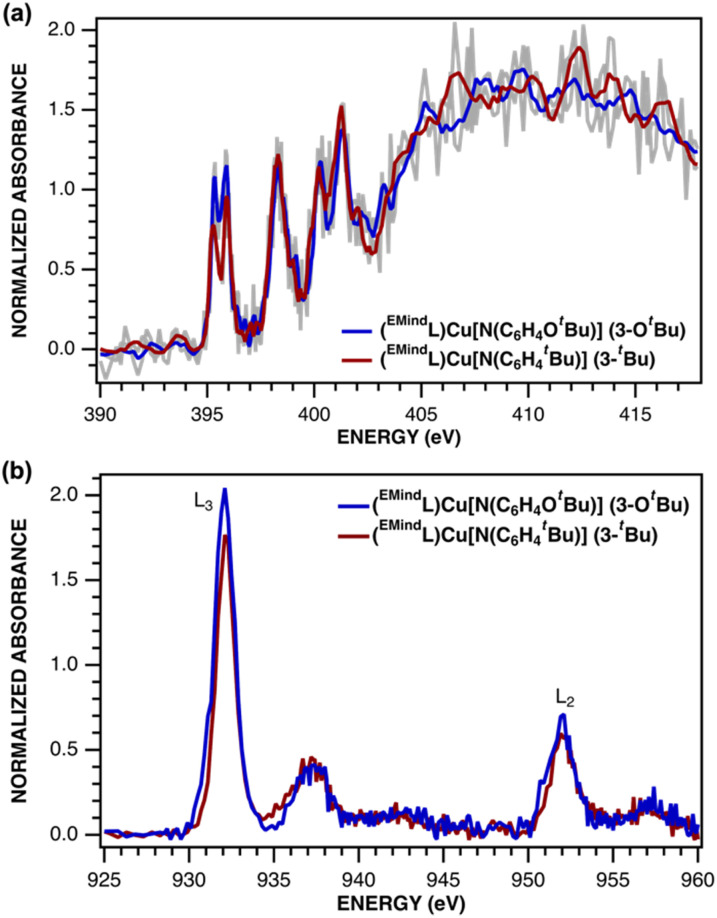
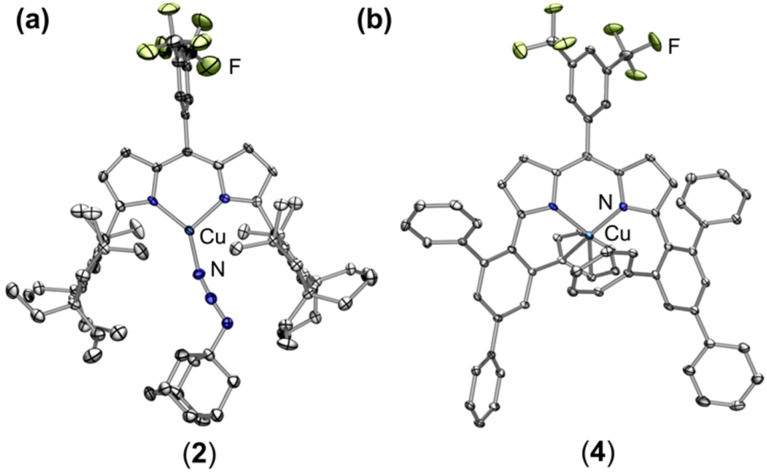
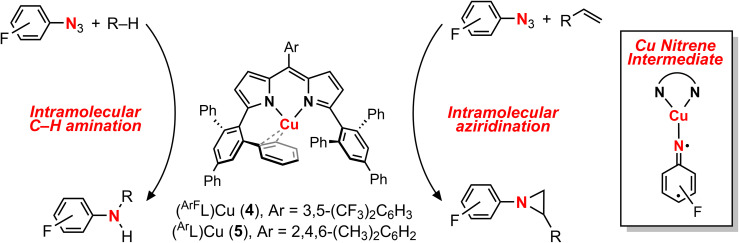
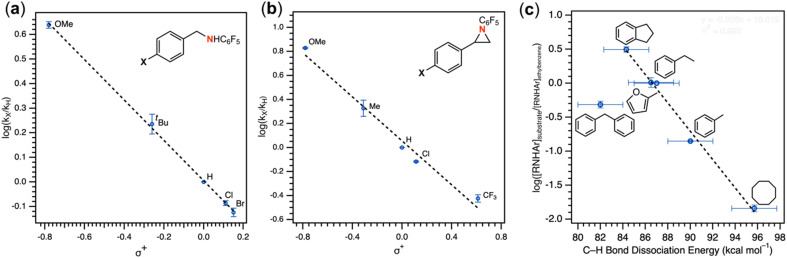
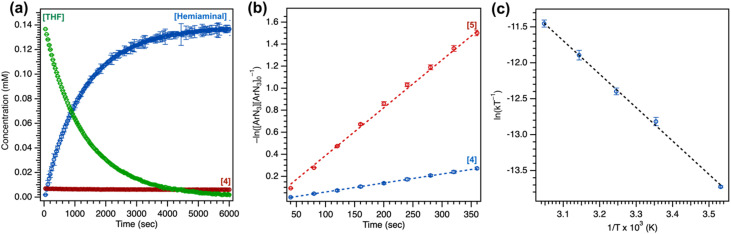
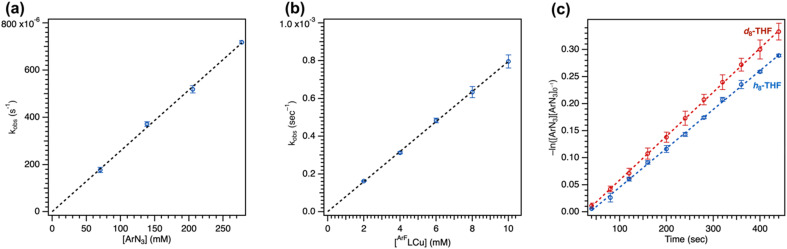
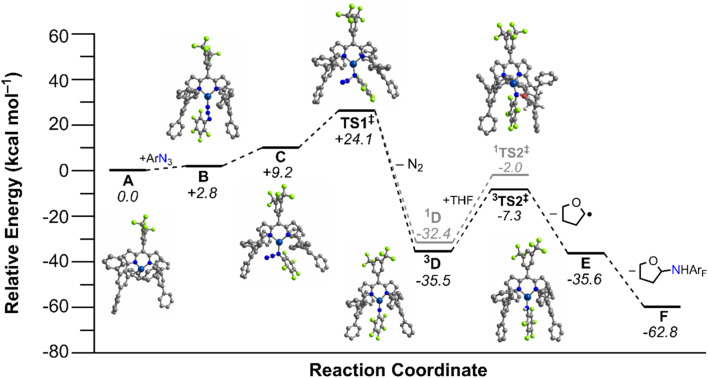
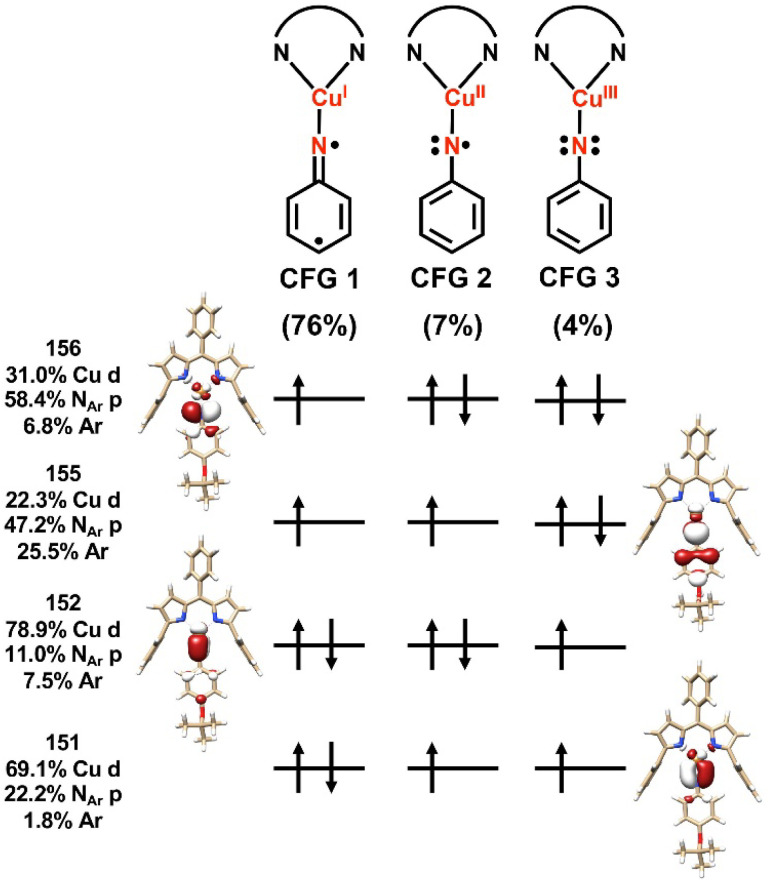
Despite the myriad Cu-catalyzed nitrene transfer methodologies to form new C-N bonds (, amination, aziridination), the critical reaction intermediates have largely eluded direct characterization due to their inherent reactivity. Herein, we report the synthesis of dipyrrin-supported Cu nitrenoid adducts, investigate their spectroscopic features, and probe their nitrene transfer chemistry through detailed mechanistic analyses. Treatment of the dipyrrin Cu complexes with substituted organoazides affords terminally ligated organoazide adducts with minimal activation of the azide unit as evidenced by vibrational spectroscopy and single crystal X-ray diffraction. The Cu nitrenoid, with an electronic structure most consistent with a triplet nitrene adduct of Cu, is accessed following geometric rearrangement of the azide adduct from κ-N terminal ligation to κ-N internal ligation with subsequent expulsion of N. For perfluorinated arylazides, stoichiometric and catalytic C-H amination and aziridination was observed. Mechanistic analysis employing substrate competition reveals an enthalpically-controlled, electrophilic nitrene transfer for primary and secondary C-H bonds. Kinetic analyses for catalytic amination using tetrahydrofuran as a model substrate reveal pseudo-first order kinetics under relevant amination conditions with a first-order dependence on both Cu and organoazide. Activation parameters determined from Eyring analysis (Δ = 9.2(2) kcal mol, Δ = -42(2) cal mol K, Δ = 21.7(2) kcal mol) and parallel kinetic isotope effect measurements (1.10(2)) are consistent with rate-limiting Cu nitrenoid formation, followed by a proposed stepwise hydrogen-atom abstraction and rapid radical recombination to furnish the resulting C-N bond. The proposed mechanism and experimental analysis are further corroborated by density functional theory calculations. Multiconfigurational calculations provide insight into the electronic structure of the catalytically relevant Cu nitrene intermediates. The findings presented herein will assist in the development of future methodology for Cu-mediated C-N bond forming catalysis.
尽管有无数种铜催化的氮烯转移方法来形成新的C-N键(如胺化、氮杂环丙烷化),但由于关键反应中间体固有的反应活性,它们的直接表征在很大程度上仍未实现。在此,我们报告了二吡咯支持的铜氮烯类加合物的合成,研究了它们的光谱特征,并通过详细的机理分析探究了它们的氮烯转移化学。用取代的有机叠氮化物处理二吡咯铜配合物,得到末端连接的有机叠氮化物加合物,振动光谱和单晶X射线衍射表明叠氮单元的活化最小。通过叠氮化物加合物从κ-N末端连接到κ-N内部连接的几何重排,随后排出N,得到电子结构与铜的三重态氮烯加合物最一致的铜氮烯类。对于全氟芳基叠氮化物,观察到了化学计量和催化的C-H胺化和氮杂环丙烷化。采用底物竞争的机理分析表明,对于一级和二级C-H键,氮烯转移是由焓控制的亲电过程。以四氢呋喃为模型底物进行催化胺化的动力学分析表明,在相关胺化条件下,反应为准一级动力学,对铜和有机叠氮化物均呈一级依赖关系。通过艾林分析确定的活化参数(Δ = 9.2(2) kcal mol,Δ = -42(2) cal mol K,Δ = 21.7(2) kcal mol)和平行动力学同位素效应测量(1.10(2))与限速的铜氮烯类形成一致,随后提出逐步氢原子提取和快速自由基重组以形成最终的C-N键。密度泛函理论计算进一步证实了所提出的机理和实验分析。多组态计算提供了对催化相关铜氮烯中间体电子结构的深入了解。本文提出的研究结果将有助于未来铜介导的C-N键形成催化方法的开发。