Hudson Institute of Medical Research, Clayton, VIC, 3168, Australia.
Department of Molecular and Translational Medicine, School of Medicine, Nursing and Health Sciences, Monash University, Clayton, VIC, 3800, Australia.
Oncogene. 2023 Nov;42(47):3529-3541. doi: 10.1038/s41388-023-02864-7. Epub 2023 Oct 16.
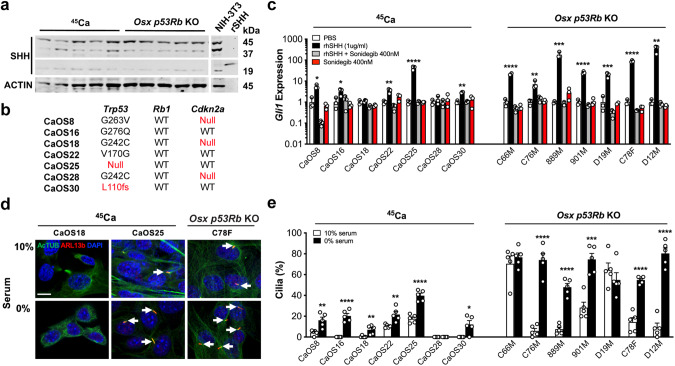
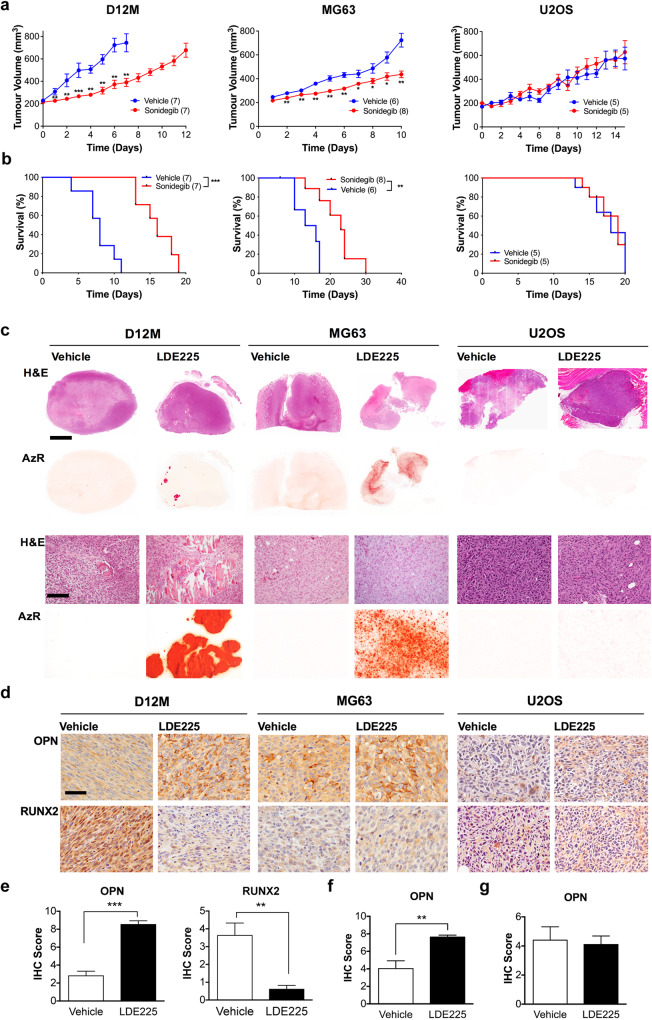
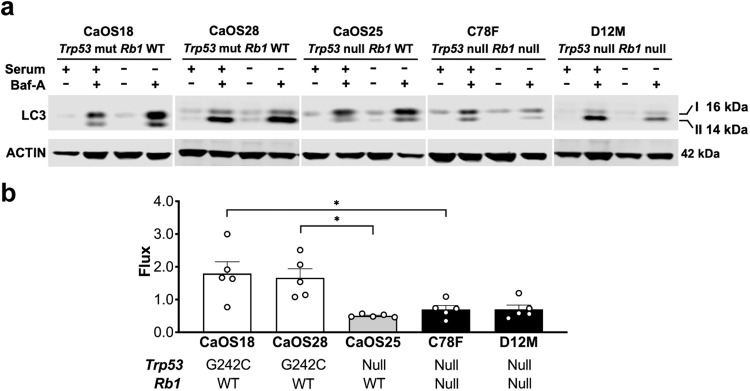
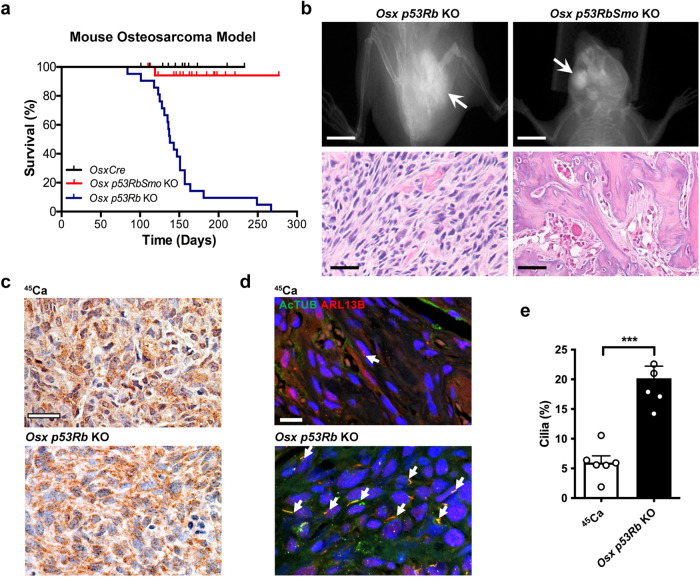
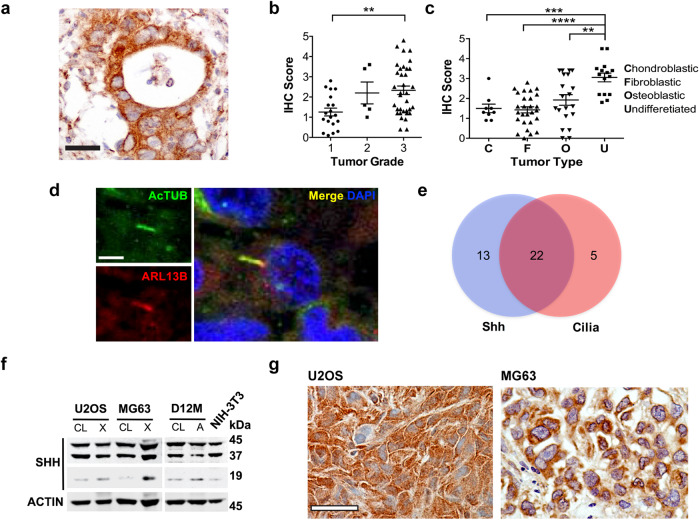
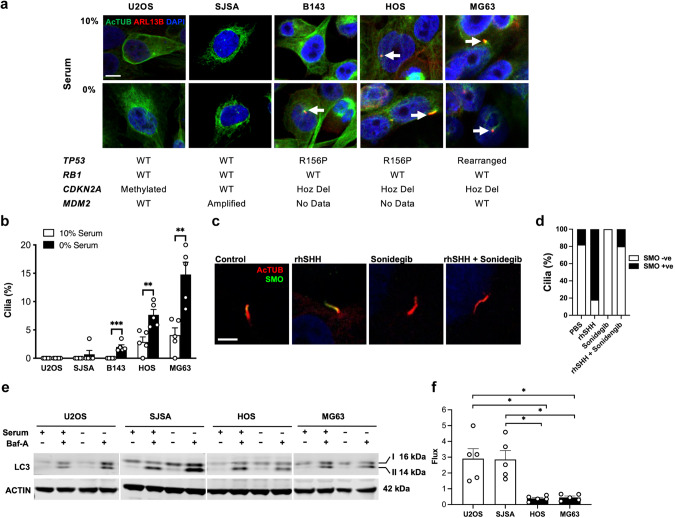
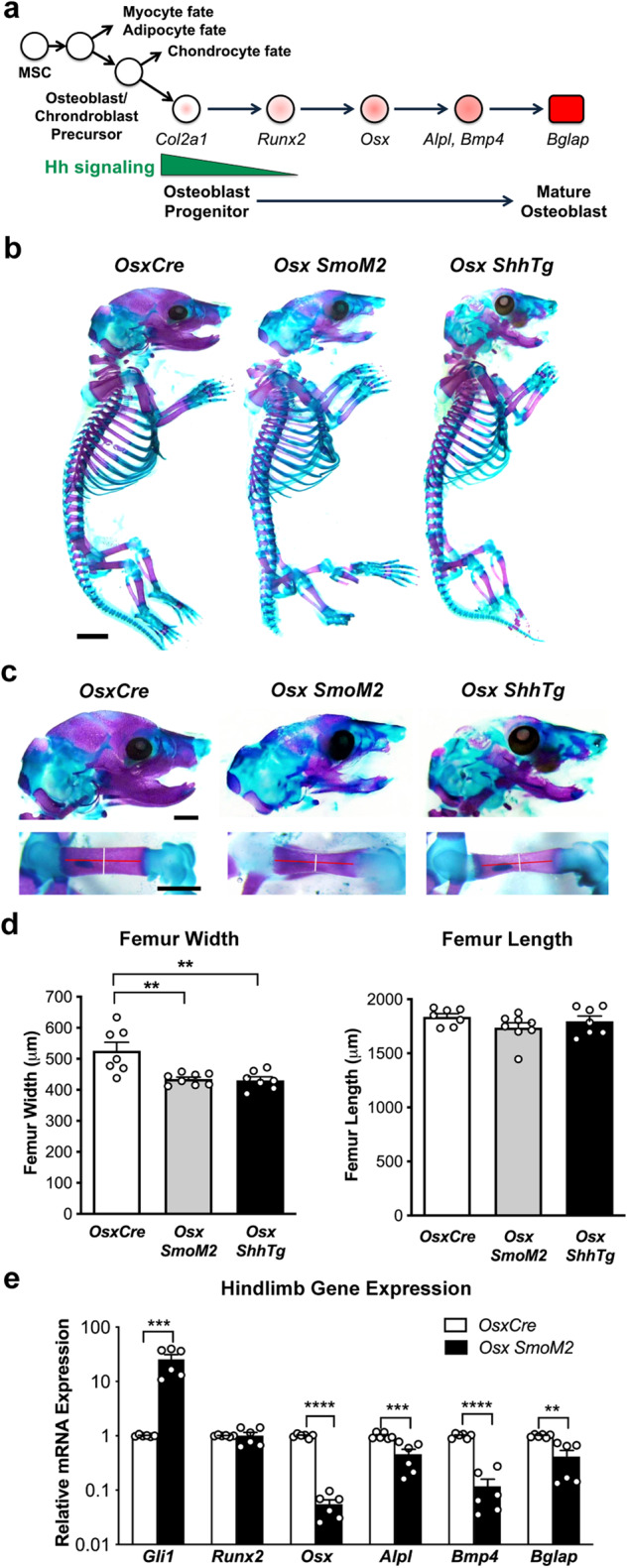
TP53 and RB1 loss-of-function mutations are common in osteosarcoma. During development, combined loss of TP53 and RB1 function leads to downregulation of autophagy and the aberrant formation of primary cilia, cellular organelles essential for the transmission of canonical Hedgehog (Hh) signaling. Excess cilia formation then leads to hypersensitivity to Hedgehog (Hh) ligand signaling. In mouse and human models, we now show that osteosarcomas with mutations in TP53 and RB1 exhibit enhanced ligand-dependent Hh pathway activation through Smoothened (SMO), a transmembrane signaling molecule required for activation of the canonical Hh pathway. This dependence is mediated by hypersensitivity to Hh ligand and is accompanied by impaired autophagy and increased primary cilia formation and expression of Hh ligand in vivo. Using a conditional genetic mouse model of Trp53 and Rb1 inactivation in osteoblast progenitors, we further show that deletion of Smo converts the highly malignant osteosarcoma phenotype to benign, well differentiated bone tumors. Conversely, conditional overexpression of SHH ligand, or a gain-of-function SMO mutant in committed osteoblast progenitors during development blocks terminal bone differentiation. Finally, we demonstrate that the SMO antagonist sonidegib (LDE225) induces growth arrest and terminal differentiation in vivo in osteosarcomas that express primary cilia and Hh ligand combined with mutations in TP53. These results provide a mechanistic framework for aberrant Hh signaling in osteosarcoma based on defining mutations in the tumor suppressor, TP53.
TP53 和 RB1 功能丧失性突变在骨肉瘤中很常见。在发育过程中,TP53 和 RB1 功能的联合缺失会导致自噬下调和初级纤毛的异常形成,而初级纤毛是经典 Hedgehog(Hh)信号转导所必需的细胞细胞器。过量的纤毛形成会导致对 Hedgehog(Hh)配体信号的过度敏感。在小鼠和人类模型中,我们现在表明,TP53 和 RB1 突变的骨肉瘤通过 Smoothened(SMO)表现出增强的配体依赖性 Hh 信号通路激活,SMO 是激活经典 Hh 信号通路所必需的跨膜信号分子。这种依赖性是通过对 Hh 配体的敏感性增加介导的,并伴有自噬受损、初级纤毛形成增加和体内 Hh 配体表达增加。使用 Trp53 和 Rb1 在成骨细胞前体细胞中失活的条件性遗传小鼠模型,我们进一步表明,Smo 的缺失将高度恶性的骨肉瘤表型转化为良性、分化良好的骨肿瘤。相反,在发育过程中,在成骨细胞前体细胞中条件性过表达 SHH 配体或功能获得性 SMO 突变会阻止终末骨分化。最后,我们证明 SMO 拮抗剂 sonidegib(LDE225)在表达初级纤毛和 Hh 配体以及 TP53 突变的骨肉瘤中诱导体内生长停滞和终末分化。这些结果为基于肿瘤抑制因子 TP53 定义突变的骨肉瘤中异常 Hh 信号提供了一个机制框架。