Institute of Molecular and Cell Biology, University of Tartu, 23 Riia Str., 51010, Tartu, Estonia.
Sci Rep. 2023 Oct 18;13(1):17765. doi: 10.1038/s41598-023-44636-z.
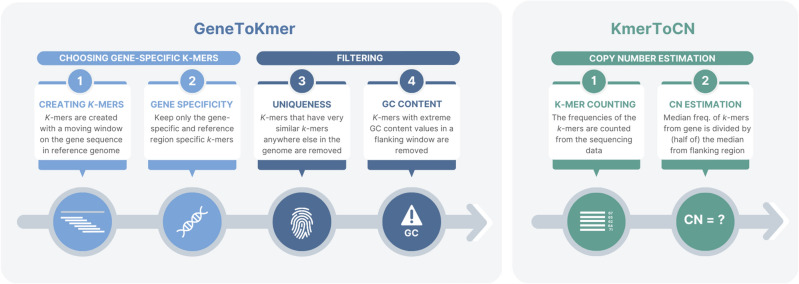
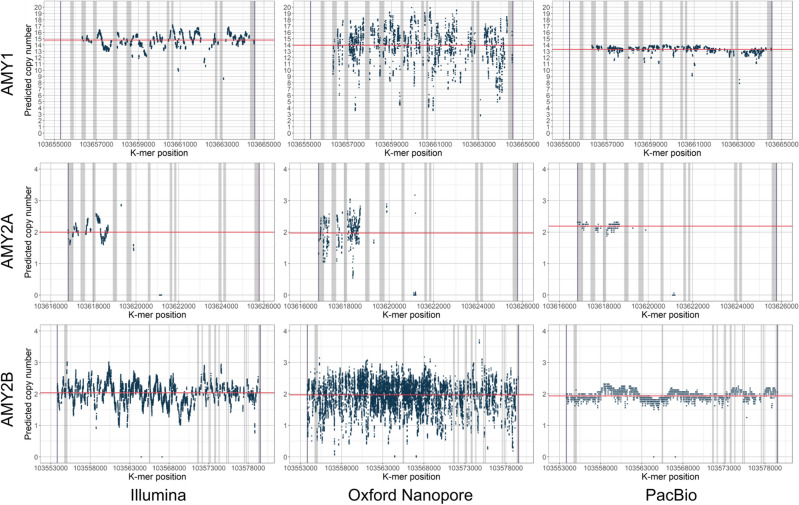
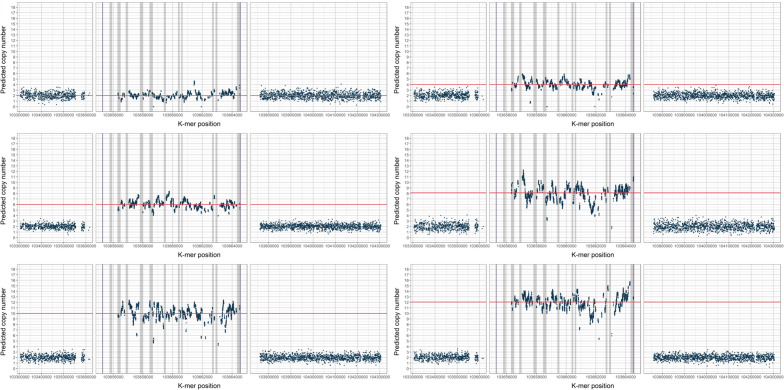
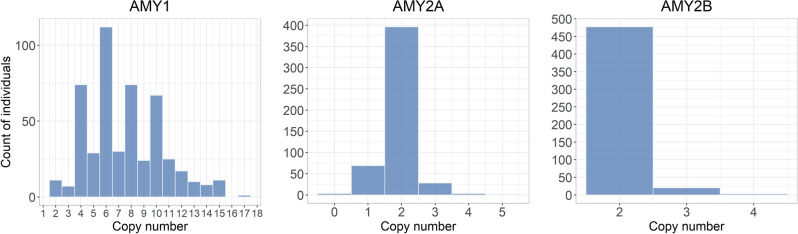
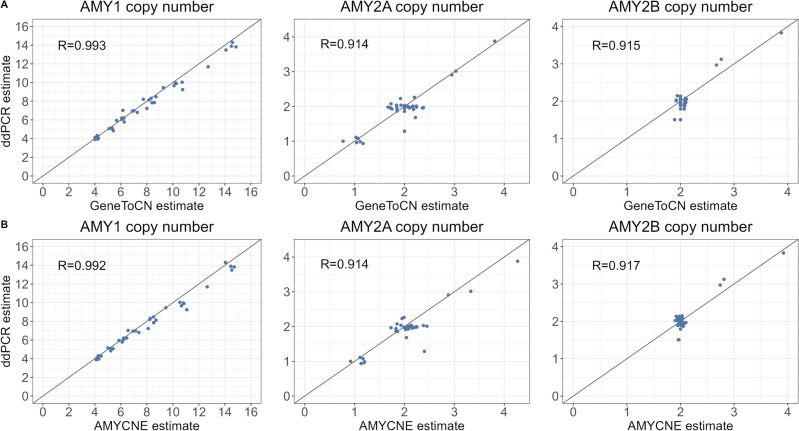
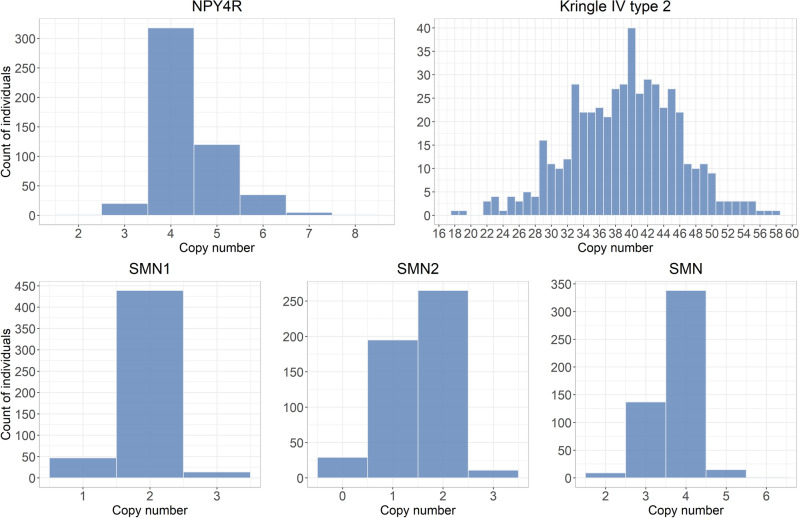
Genomes exhibit large regions with segmental copy number variation, many of which include entire genes and are multiallelic. We have developed a computational method GeneToCN that counts the frequencies of gene-specific k-mers in FASTQ files and uses this information to infer copy number of the gene. We validated the copy number predictions for amylase genes (AMY1, AMY2A, AMY2B) using experimental data from digital droplet PCR (ddPCR) on 39 individuals and observed a strong correlation (R = 0.99) between GeneToCN predictions and experimentally determined copy numbers. An additional validation on FCGR3 genes showed a higher concordance for FCGR3A compared to two other methods, but reduced accuracy for FCGR3B. We further tested the method on three different genomic regions (SMN, NPY4R, and LPA Kringle IV-2 domain). Predicted copy number distributions of these genes in a set of 500 individuals from the Estonian Biobank were in good agreement with the previously published studies. In addition, we investigated the possibility to use GeneToCN on sequencing data generated by different technologies by comparing copy number predictions from Illumina, PacBio, and Oxford Nanopore data of the same sample. Despite the differences in variability of k-mer frequencies, all three sequencing technologies give similar predictions with GeneToCN.
基因组表现出具有片段拷贝数变异的大片段区域,其中许多区域包括整个基因且具有多等位基因。我们开发了一种计算方法 GeneToCN,它可以计算 FASTQ 文件中基因特异性 k-mer 的频率,并利用这些信息推断基因的拷贝数。我们使用来自 39 个人的数字液滴 PCR (ddPCR) 的实验数据验证了淀粉酶基因 (AMY1、AMY2A、AMY2B) 的拷贝数预测,发现 GeneToCN 预测和实验确定的拷贝数之间存在很强的相关性 (R = 0.99)。对 FCGR3 基因的进一步验证表明,与其他两种方法相比,FCGR3A 的一致性更高,但 FCGR3B 的准确性降低。我们还在三个不同的基因组区域 (SMN、NPY4R 和 LPA kringle IV-2 结构域) 上测试了该方法。在来自爱沙尼亚生物库的 500 个人的一组中,这些基因的预测拷贝数分布与之前发表的研究结果非常一致。此外,我们通过比较相同样本的 Illumina、PacBio 和 Oxford Nanopore 数据的拷贝数预测,研究了使用不同技术生成的测序数据GeneToCN 的可能性。尽管 k-mer 频率的可变性存在差异,但所有三种测序技术都可以使用 GeneToCN 给出相似的预测。