Vanderbilt Genetics Institute, Vanderbilt University School of Medicine, Nashville, TN, 37232, USA.
Department of Biochemistry, Vanderbilt University School of Medicine, Nashville, TN, 37232, USA.
BMC Genomics. 2023 Oct 19;24(1):623. doi: 10.1186/s12864-023-09622-9.
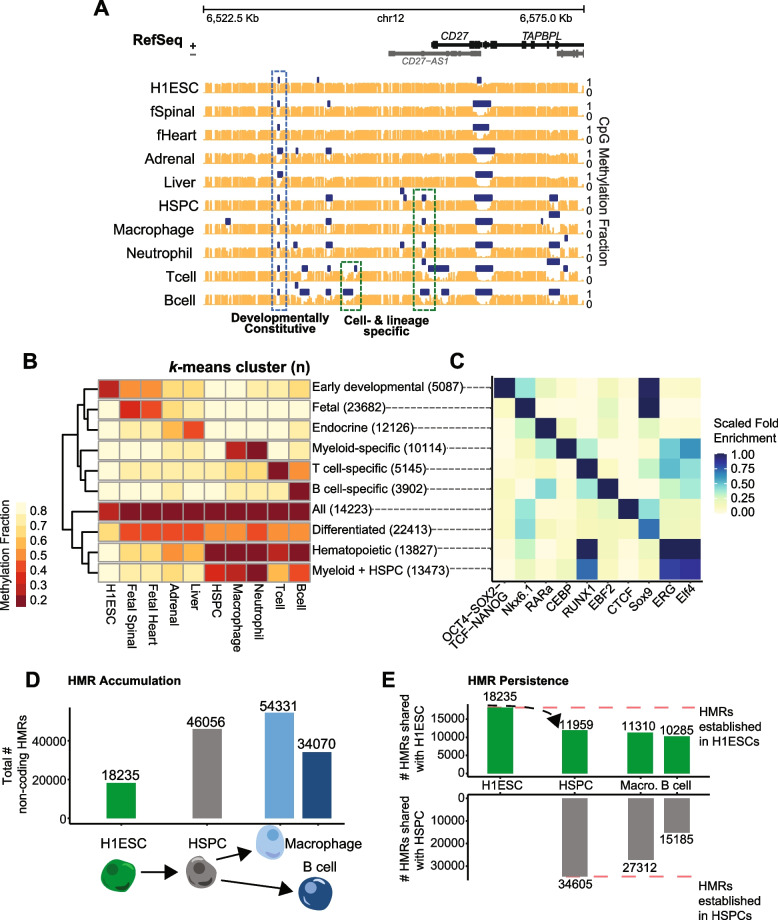
Establishment of DNA methylation (DNAme) patterns is essential for balanced multi-lineage cellular differentiation, but exactly how these patterns drive cellular phenotypes is unclear. While > 80% of CpG sites are stably methylated, tens of thousands of discrete CpG loci form hypomethylated regions (HMRs). Because they lack DNAme, HMRs are considered transcriptionally permissive, but not all HMRs actively regulate genes. Unlike promoter HMRs, a subset of non-coding HMRs is cell type-specific and enriched for tissue-specific gene regulatory functions. Our data further argues not only that HMR establishment is an important step in enforcing cell identity, but also that cross-cell type and spatial HMR patterns are functionally informative of gene regulation.
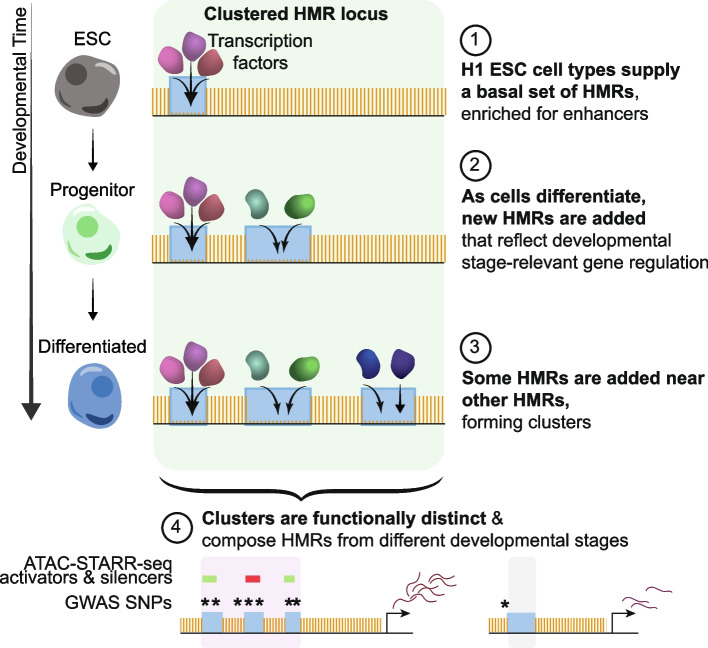
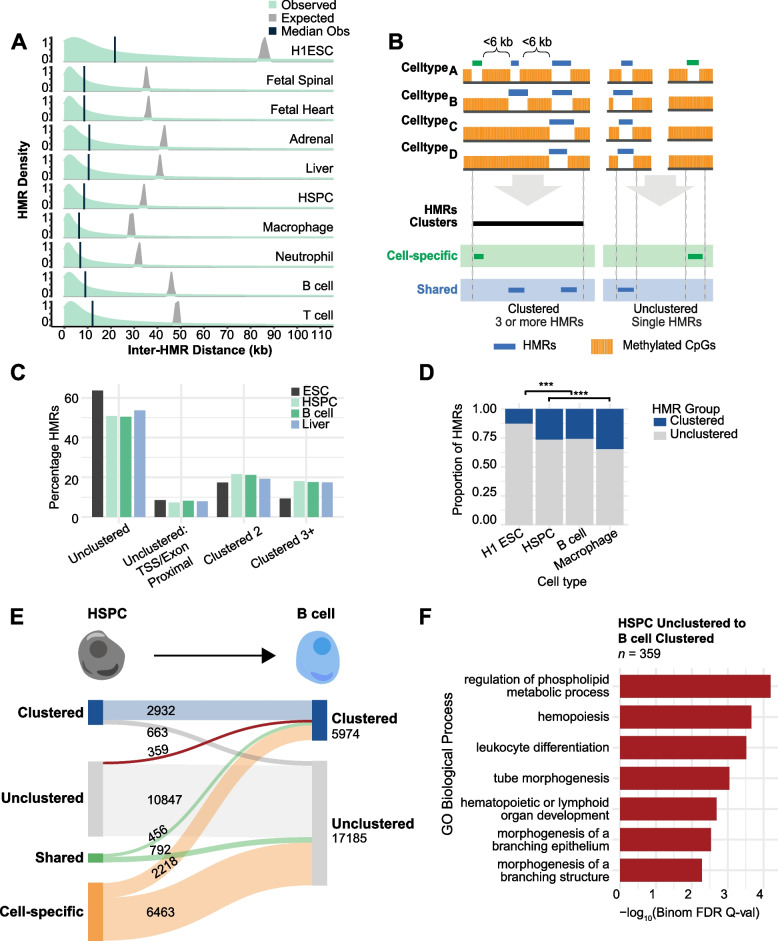
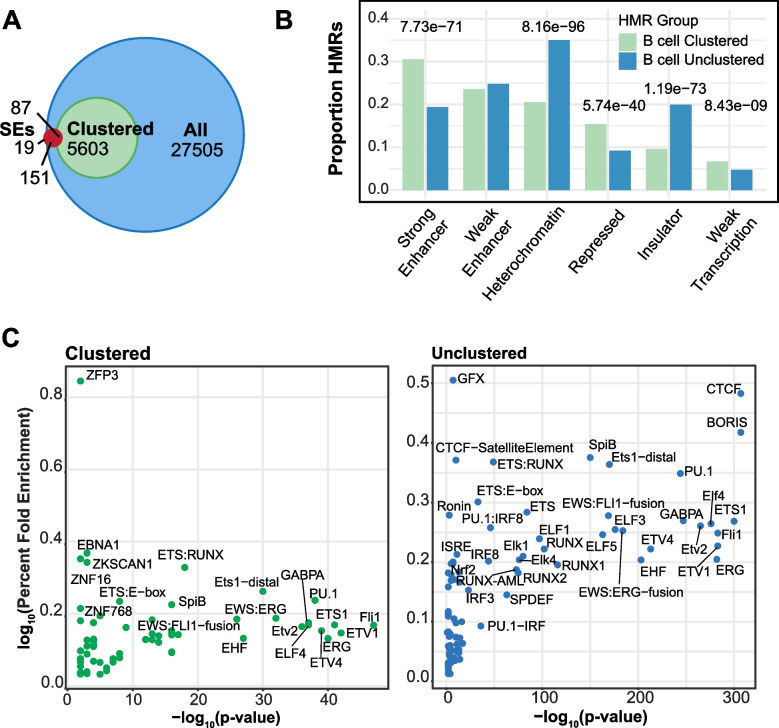
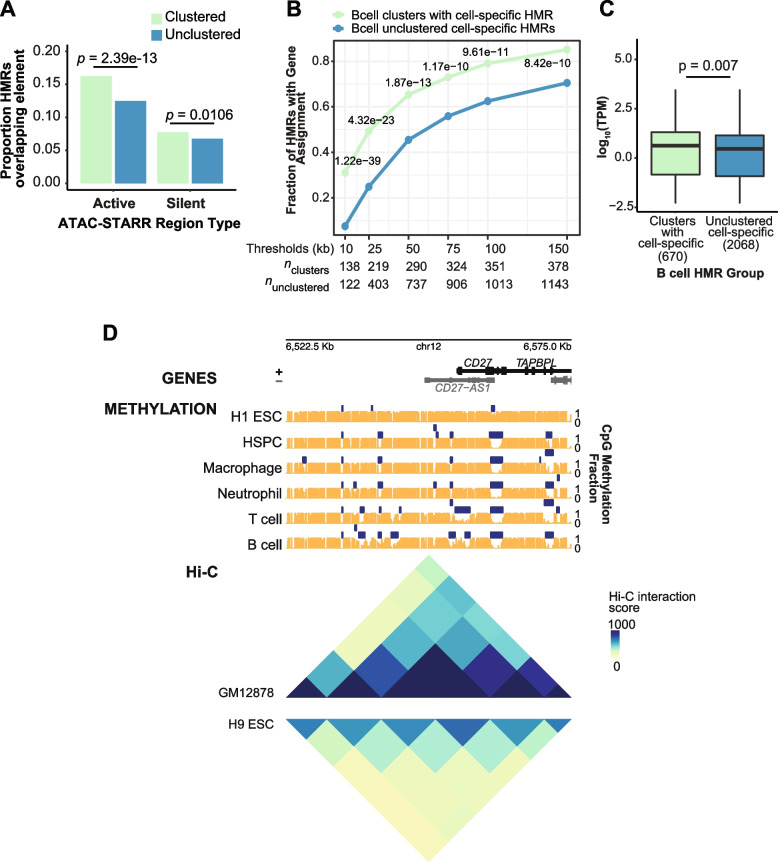
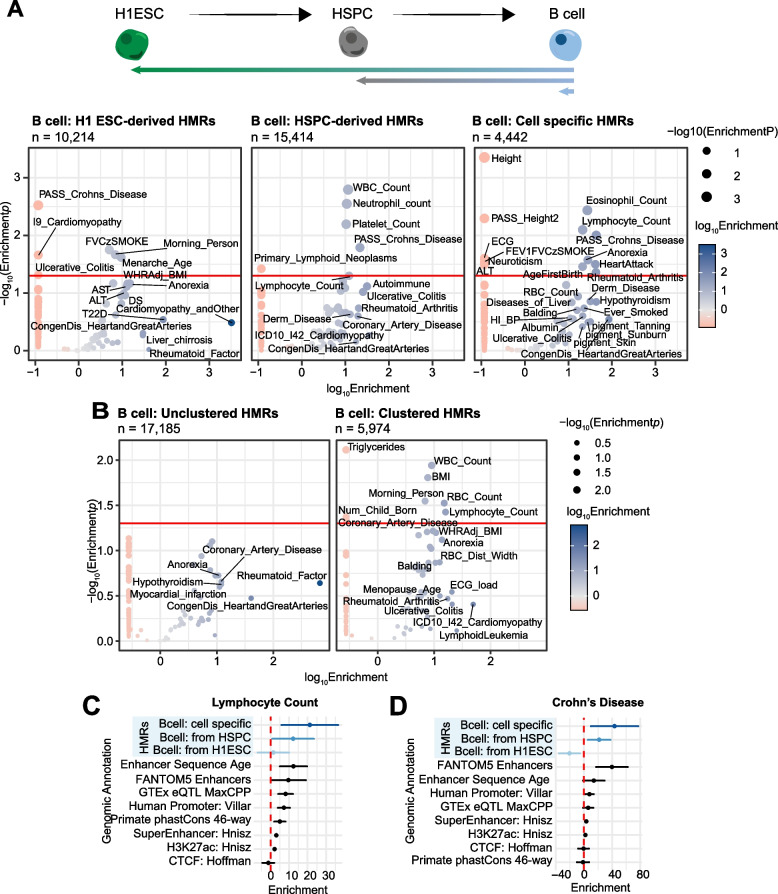
To understand the significance of non-coding HMRs, we systematically dissected HMR patterns across diverse human cell types and developmental timepoints, including embryonic, fetal, and adult tissues. Unsupervised clustering of 126,104 distinct HMRs revealed that levels of HMR specificity reflects a developmental hierarchy supported by enrichment of stage-specific transcription factors and gene ontologies. Using a pseudo-time course of development from embryonic stem cells to adult stem and mature hematopoietic cells, we find that most HMRs observed in differentiated cells (~ 60%) are established at early developmental stages and accumulate as development progresses. HMRs that arise during differentiation frequently (~ 35%) establish near existing HMRs (≤ 6 kb away), leading to the formation of HMR clusters associated with stronger enhancer activity. Using SNP-based partitioned heritability from GWAS summary statistics across diverse traits and clinical lab values, we discovered that genetic contribution to trait heritability is enriched within HMRs. Moreover, the contribution of heritability to cell-relevant traits increases with both increasing HMR specificity and HMR clustering, supporting the role of distinct HMR subsets in regulating normal cell function.
Our results demonstrate that the entire HMR repertoire within a cell-type, rather than just the cell type-specific HMRs, stores information that is key to understanding and predicting cellular phenotypes. Ultimately, these data provide novel insights into how DNA hypo-methylation provides genetically distinct historical records of a cell's journey through development, highlighting HMRs as functionally distinct from other epigenomic annotations.
DNA 甲基化(DNAme)模式的建立对于平衡的多谱系细胞分化至关重要,但这些模式如何驱动细胞表型尚不清楚。虽然>80%的 CpG 位点稳定甲基化,但数以万计的离散 CpG 位点形成低甲基化区域(HMR)。由于它们缺乏 DNAme,HMR 被认为是转录允许的,但并非所有 HMR 都主动调节基因。与启动子 HMR 不同,一组非编码 HMR 是细胞类型特异性的,并且富含组织特异性基因调控功能。我们的数据进一步证明,HMR 的建立不仅是强化细胞身份的重要步骤,而且跨细胞类型和空间的 HMR 模式在基因调控方面具有功能信息。
为了了解非编码 HMR 的意义,我们系统地剖析了人类不同细胞类型和发育时间点的 HMR 模式,包括胚胎、胎儿和成人组织。对 126104 个不同 HMR 的无监督聚类显示,HMR 特异性的水平反映了一个由阶段特异性转录因子和基因本体论支持的发育层次结构。使用从胚胎干细胞到成人干细胞和成熟造血细胞的发育伪时间过程,我们发现分化细胞中观察到的大多数 HMR(60%)是在早期发育阶段建立的,并随着发育的进行而积累。在分化过程中出现的 HMR 经常(35%)在现有 HMR 附近(≤6 kb 以内)建立,导致与更强的增强子活性相关的 HMR 簇的形成。利用来自不同性状和临床实验室值的 GWAS 汇总统计数据的 SNP 基分位数遗传力,我们发现,遗传对性状遗传力的贡献在 HMR 中富集。此外,遗传对与细胞相关的性状的贡献随着 HMR 特异性和 HMR 聚类的增加而增加,这支持了不同 HMR 亚类在调节正常细胞功能方面的作用。
我们的结果表明,细胞类型内的整个 HMR 库,而不仅仅是细胞类型特异性的 HMR,存储了理解和预测细胞表型的关键信息。最终,这些数据为 DNA 低甲基化如何为细胞发育过程中的细胞旅程提供遗传上独特的历史记录提供了新的见解,突出了 HMR 与其他表观遗传注释的功能区别。