Chatterjee Raghunath, He Ximiao, Huang Di, FitzGerald Peter, Smith Andrew, Vinson Charles
Laboratory of Metabolism, National Cancer Institute, National Institutes of Health, 37 Convent Drive, Bethesda, MD 20892 USA.
Human Genetics Unit, Indian Statistical Institute, 203 B. T. Road, Kolkata, 700108 India.
Epigenetics Chromatin. 2014 Dec 2;7:35. doi: 10.1186/1756-8935-7-35. eCollection 2014.
Genome-wide DNA methylation at a single nucleotide resolution in different primary cells of the mammalian genome helps to determine the characteristics and functions of tissue-specific hypomethylated regions (TS-HMRs). We determined genome-wide cytosine methylation maps at 91X and 36X coverage of newborn female mouse primary dermal fibroblasts and keratinocytes and compared with mRNA-seq gene expression data.
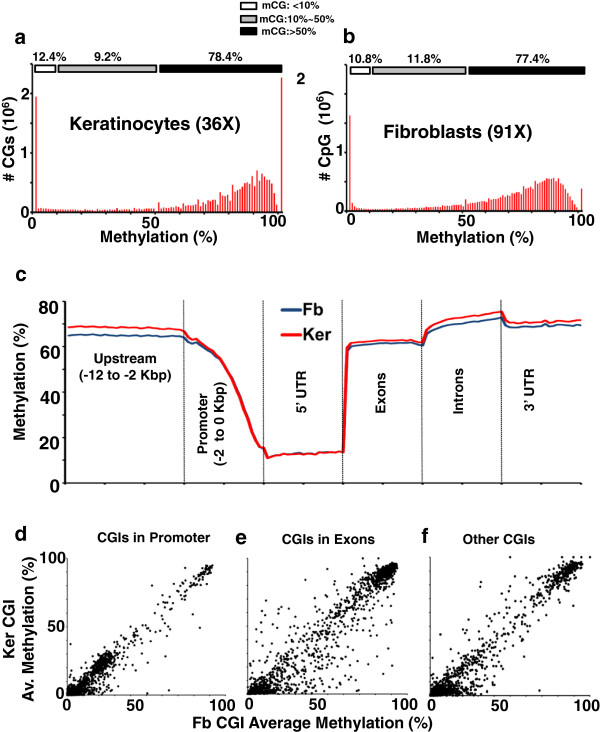
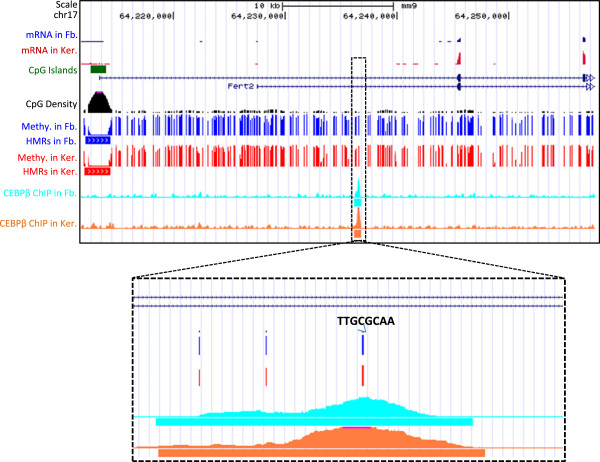
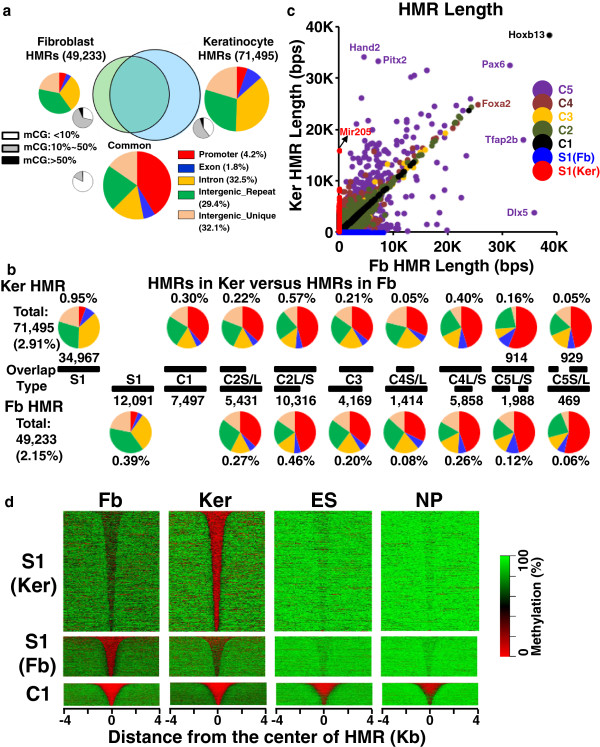
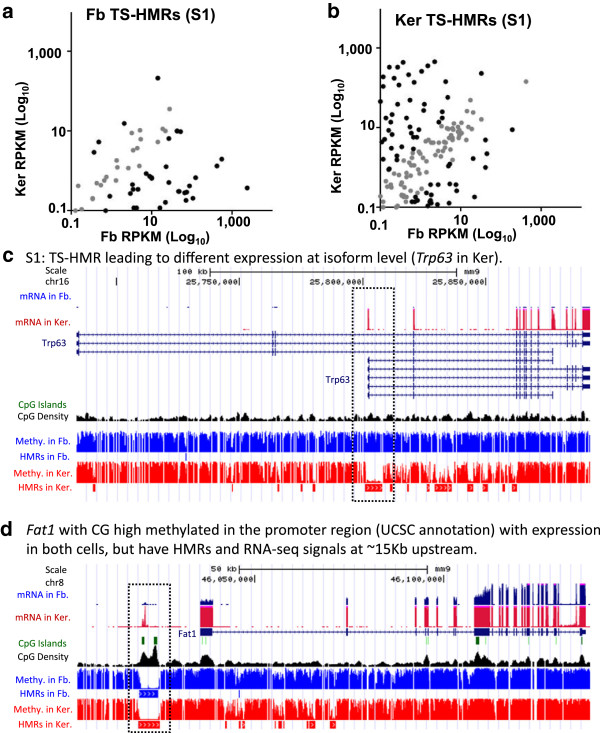
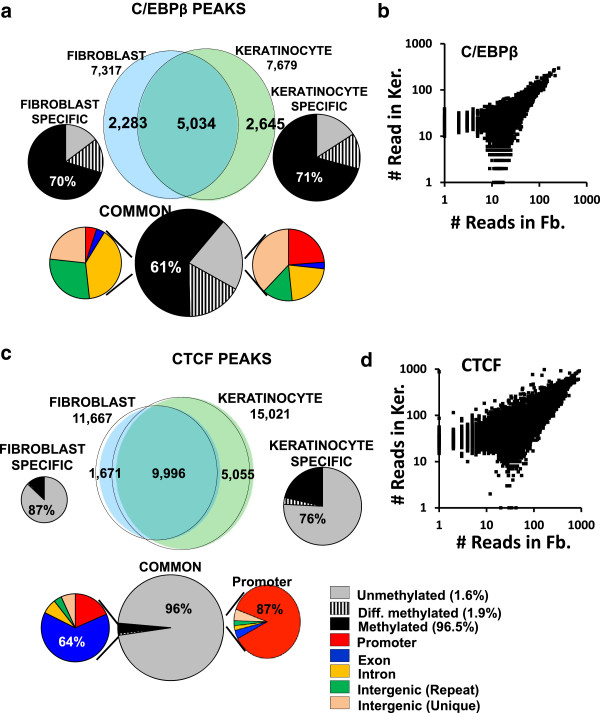
These high coverage methylation maps were used to identify HMRs in both cell types. A total of 2.91% of the genome are in keratinocyte HMRs, and 2.15% of the genome are in fibroblast HMRs with 1.75% being common. Half of the TS-HMRs are extensions of common HMRs, and the remaining are unique TS-HMRs. Four levels of CG methylation are observed: 1) total unmethylation for CG dinucleotides in HMRs in CGIs that are active in all tissues; 2) 10% to 40% methylation for TS-HMRs; 3) 60% methylation for TS-HMRs in cells types where they are not in HMRs; and 4) 70% methylation for the nonfunctioning part of the genome. SINE elements are depleted inside the TS-HMRs, while highly enriched in the surrounding regions. Hypomethylation at the last exon shows gene repression, while demethylation toward the gene body positively correlates with gene expression. The overlapping HMRs have a more complex relationship with gene expression. The common HMRs and TS-HMRs are each enriched for distinct Transcription Factor Binding Sites (TFBS). C/EBPβ binds to methylated regions outside of HMRs while CTCF prefers to bind in HMRs, highlighting these two parts of the genome and their potential interactions.
Keratinocytes and fibroblasts are of epithelial and mesenchymal origin. High-resolution methylation maps in these two cell types can be used as reference methylomes for analyzing epigenetic mechanisms in several diseases including cancer. Please see related article at the following link: http://www.epigeneticsandchromatin.com/content/7/1/34.
哺乳动物基因组中不同原代细胞的单核苷酸分辨率全基因组DNA甲基化有助于确定组织特异性低甲基化区域(TS-HMR)的特征和功能。我们确定了新生雌性小鼠原代表皮成纤维细胞和角质形成细胞在91X和36X覆盖度下的全基因组胞嘧啶甲基化图谱,并与mRNA测序基因表达数据进行了比较。
这些高覆盖度甲基化图谱用于鉴定两种细胞类型中的HMR。角质形成细胞HMRs占基因组的2.91%,成纤维细胞HMRs占基因组的2.15%,其中1.75%是共同的。一半的TS-HMRs是共同HMRs的延伸,其余是独特的TS-HMRs。观察到四种水平的CG甲基化:1)在所有组织中都有活性的CGI中的HMRs中CG二核苷酸的完全未甲基化;2)TS-HMRs的甲基化水平为10%至40%;3)在非HMRs的细胞类型中TS-HMRs的甲基化水平为60%;4)基因组无功能部分的甲基化水平为70%。SINE元件在TS-HMRs内部缺失,而在周围区域高度富集。最后一个外显子的低甲基化显示基因抑制,而向基因体的去甲基化与基因表达呈正相关。重叠的HMRs与基因表达有更复杂的关系。共同HMRs和TS-HMRs各自富集了不同的转录因子结合位点(TFBS)。C/EBPβ与HMRs外部的甲基化区域结合,而CTCF更喜欢结合在HMRs中,突出了基因组的这两个部分及其潜在相互作用。
角质形成细胞和成纤维细胞分别起源于上皮和间充质。这两种细胞类型的高分辨率甲基化图谱可作为参考甲基化组,用于分析包括癌症在内的多种疾病的表观遗传机制。请点击以下链接查看相关文章:http://www.epigeneticsandchromatin.com/content/7/1/34。