Menzel Michael, Ossowski Stephan, Kral Sebastian, Metzger Patrick, Horak Peter, Marienfeld Ralf, Boerries Melanie, Wolter Steffen, Ball Markus, Neumann Olaf, Armeanu-Ebinger Sorin, Schroeder Christopher, Matysiak Uta, Goldschmid Hannah, Schipperges Vincent, Fürstberger Axel, Allgäuer Michael, Eberhardt Timo, Niewöhner Jakob, Blaumeiser Andreas, Ploeger Carolin, Haack Tobias Bernd, Tay Timothy Kwang Yong, Kelemen Olga, Pauli Thomas, Kirchner Martina, Kluck Klaus, Ott Alexander, Renner Marcus, Admard Jakob, Gschwind Axel, Lassmann Silke, Kestler Hans, Fend Falko, Illert Anna Lena, Werner Martin, Möller Peter, Seufferlein Thomas Theodor Werner, Malek Nisar, Schirmacher Peter, Fröhling Stefan, Kazdal Daniel, Budczies Jan, Stenzinger Albrecht
Institute of Pathology, Heidelberg University Hospital, Heidelberg, Germany.
Center for Personalized Medicine (ZPM), Heidelberg, Germany.
NPJ Precis Oncol. 2023 Oct 20;7(1):106. doi: 10.1038/s41698-023-00457-x.
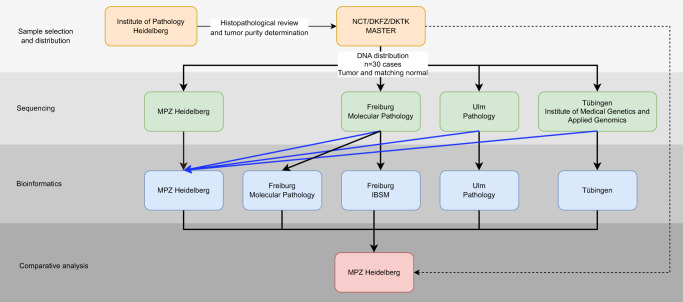
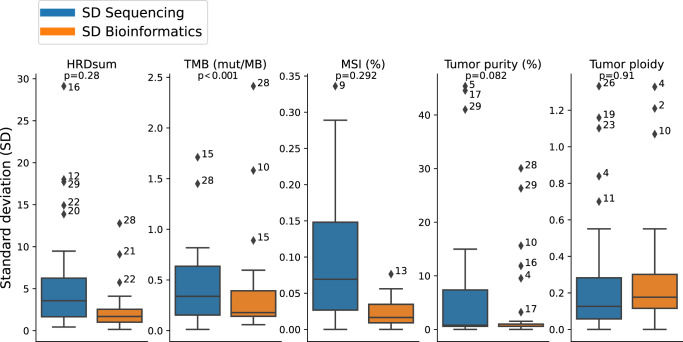
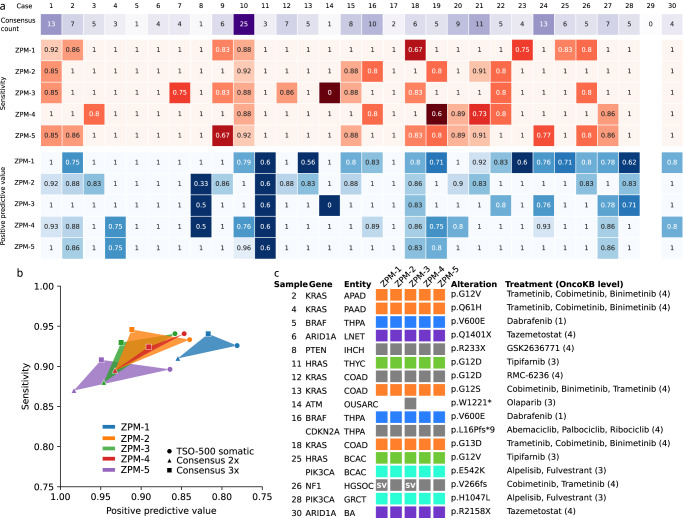
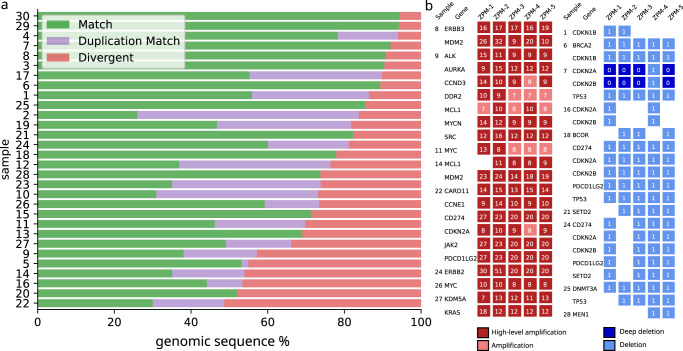
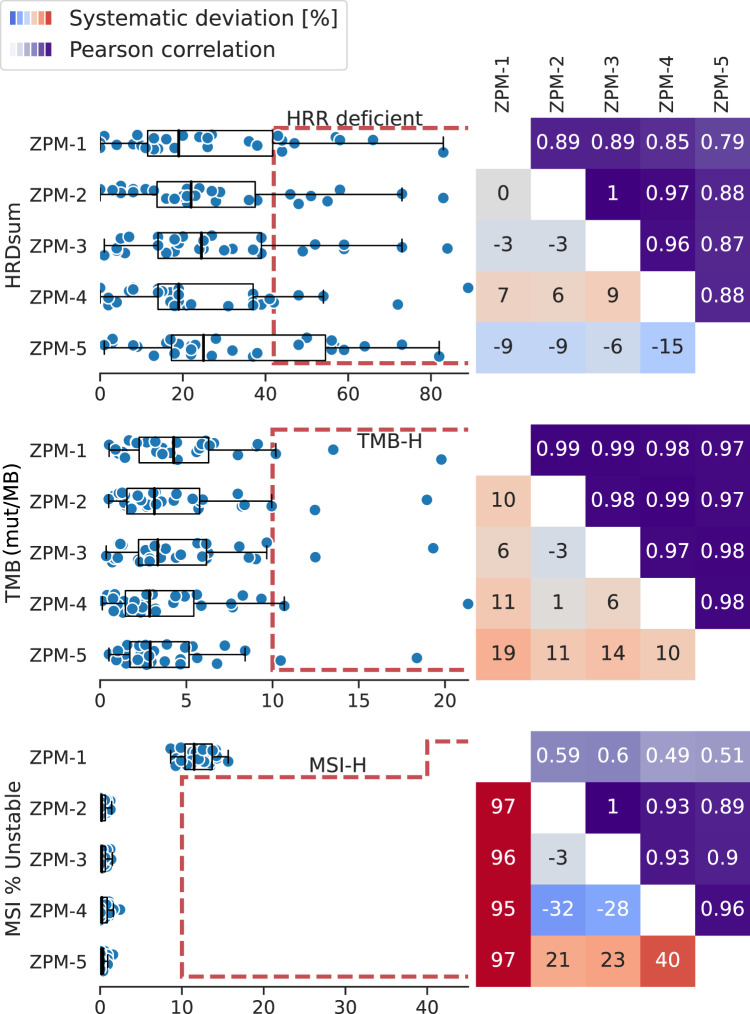
A growing number of druggable targets and national initiatives for precision oncology necessitate broad genomic profiling for many cancer patients. Whole exome sequencing (WES) offers unbiased analysis of the entire coding sequence, segmentation-based detection of copy number alterations (CNAs), and accurate determination of complex biomarkers including tumor mutational burden (TMB), homologous recombination repair deficiency (HRD), and microsatellite instability (MSI). To assess the inter-institution variability of clinical WES, we performed a comparative pilot study between German Centers of Personalized Medicine (ZPMs) from five participating institutions. Tumor and matched normal DNA from 30 patients were analyzed using custom sequencing protocols and bioinformatic pipelines. Calling of somatic variants was highly concordant with a positive percentage agreement (PPA) between 91 and 95% and a positive predictive value (PPV) between 82 and 95% compared with a three-institution consensus and full agreement for 16 of 17 druggable targets. Explanations for deviations included low VAF or coverage, differing annotations, and different filter protocols. CNAs showed overall agreement in 76% for the genomic sequence with high wet-lab variability. Complex biomarkers correlated strongly between institutions (HRD: 0.79-1, TMB: 0.97-0.99) and all institutions agreed on microsatellite instability. This study will contribute to the development of quality control frameworks for comprehensive genomic profiling and sheds light onto parameters that require stringent standardization.
越来越多可成药靶点以及精准肿瘤学国家倡议使得许多癌症患者需要进行广泛的基因组分析。全外显子组测序(WES)能够对整个编码序列进行无偏分析、基于分割的拷贝数变异(CNA)检测,以及准确测定包括肿瘤突变负荷(TMB)、同源重组修复缺陷(HRD)和微卫星不稳定性(MSI)在内的复杂生物标志物。为了评估临床WES的机构间变异性,我们在来自五个参与机构的德国个性化医学中心(ZPMs)之间开展了一项比较性试点研究。使用定制测序方案和生物信息学流程对30例患者的肿瘤及配对正常DNA进行分析。与三机构共识相比,体细胞变异的检出高度一致,阳性百分比一致性(PPA)在91%至95%之间,阳性预测值(PPV)在82%至95%之间,并且对于17个可成药靶点中的16个完全一致。偏差的原因包括低变异等位基因频率(VAF)或覆盖度、不同注释以及不同过滤方案。对于基因组序列,CNA总体一致性为76%,实验室内变异性较高。各机构之间复杂生物标志物的相关性很强(HRD:0.79 - 1,TMB:0.97 - 0.99),并且所有机构在微卫星不稳定性方面达成一致。本研究将有助于全面基因组分析质量控制框架的开发,并揭示需要严格标准化的参数。