Tiwari Ratnakar, Sharma Rajni, Rajendran Ganeshkumar, Borkowski Gabriella S, An Si Young, Schonfeld Michael, O'Sullivan James, Schipma Matthew J, Zhou Yalu, Courbon Guillaume, David Valentin, Quaggin Susan E, Thorp Edward, Chandel Navdeep S, Kapitsinou Pinelopi P
Feinberg Cardiovascular Research Institute, Northwestern University Feinberg School of Medicine, Chicago, IL.
Division of Nephrology & Hypertension, Northwestern University Feinberg School of Medicine, Chicago, IL, USA.
bioRxiv. 2023 Oct 3:2023.10.03.560700. doi: 10.1101/2023.10.03.560700.
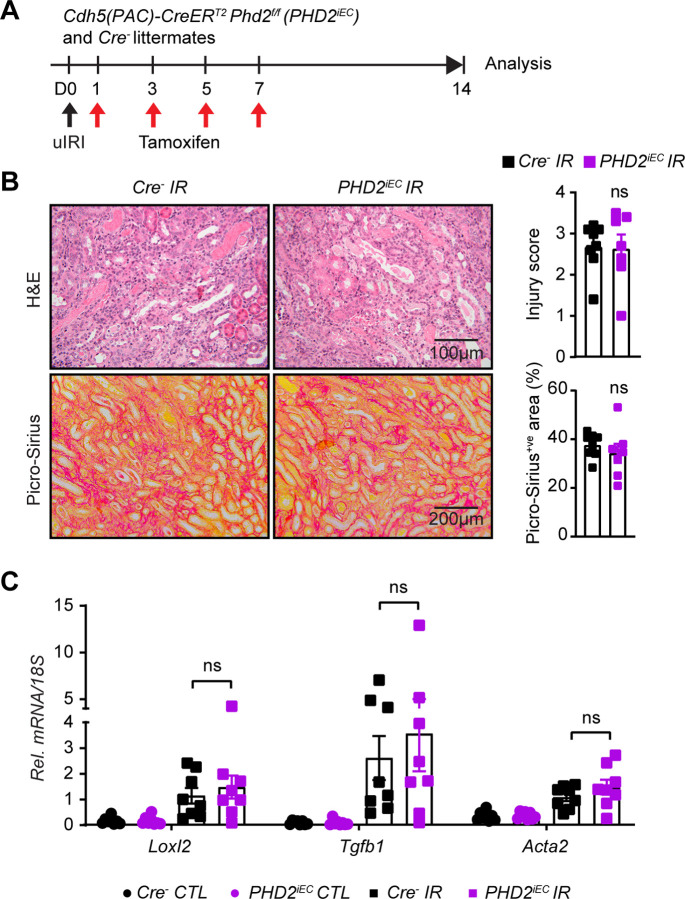
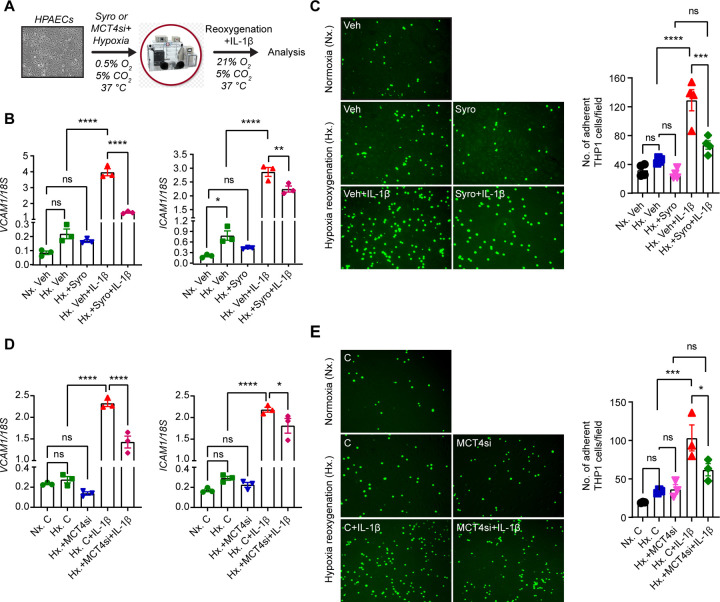
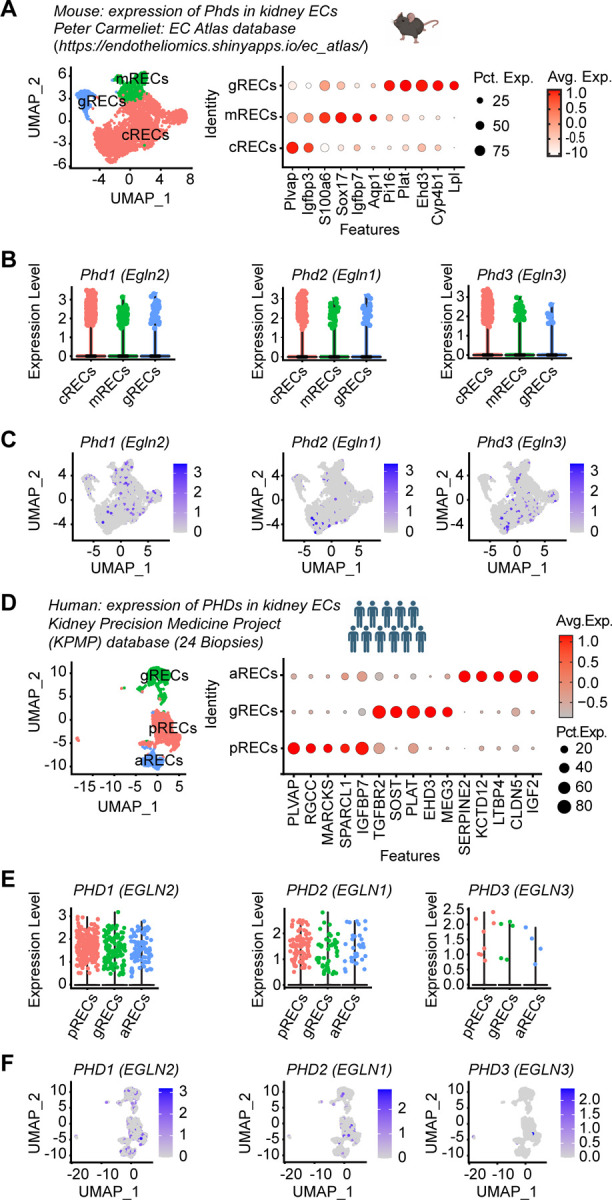
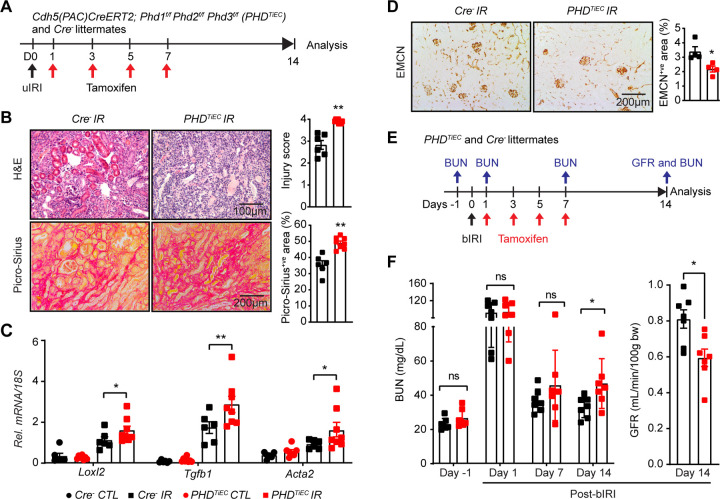
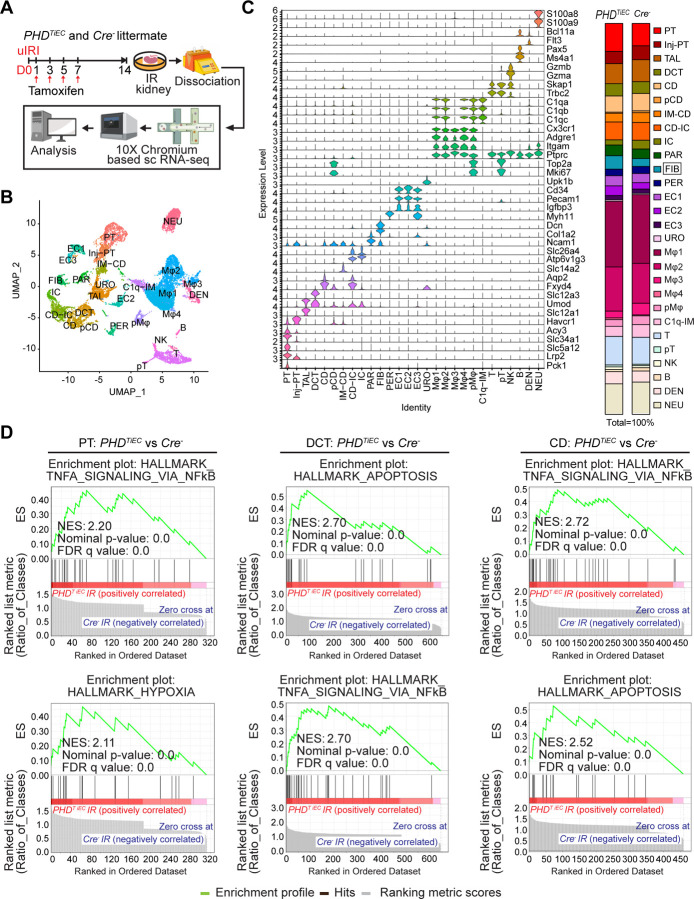
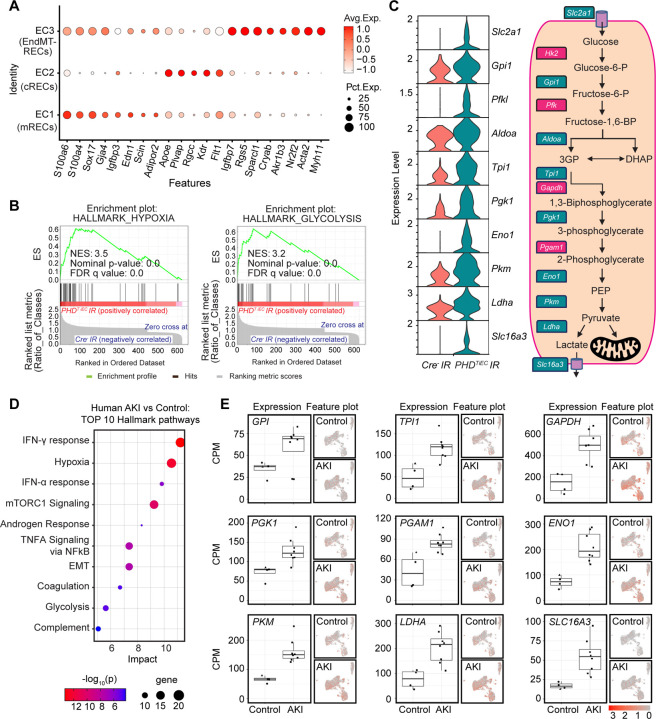
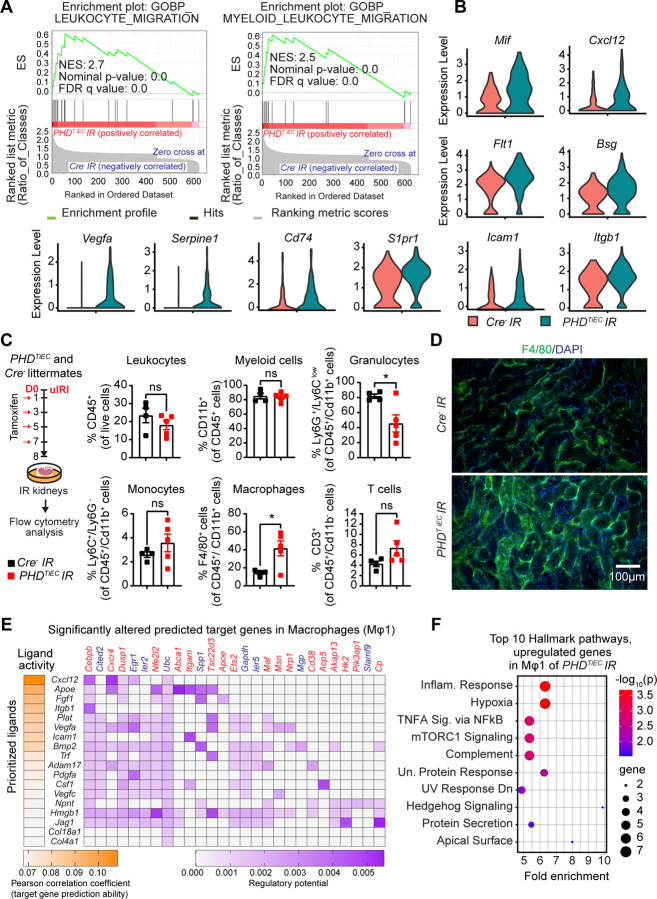
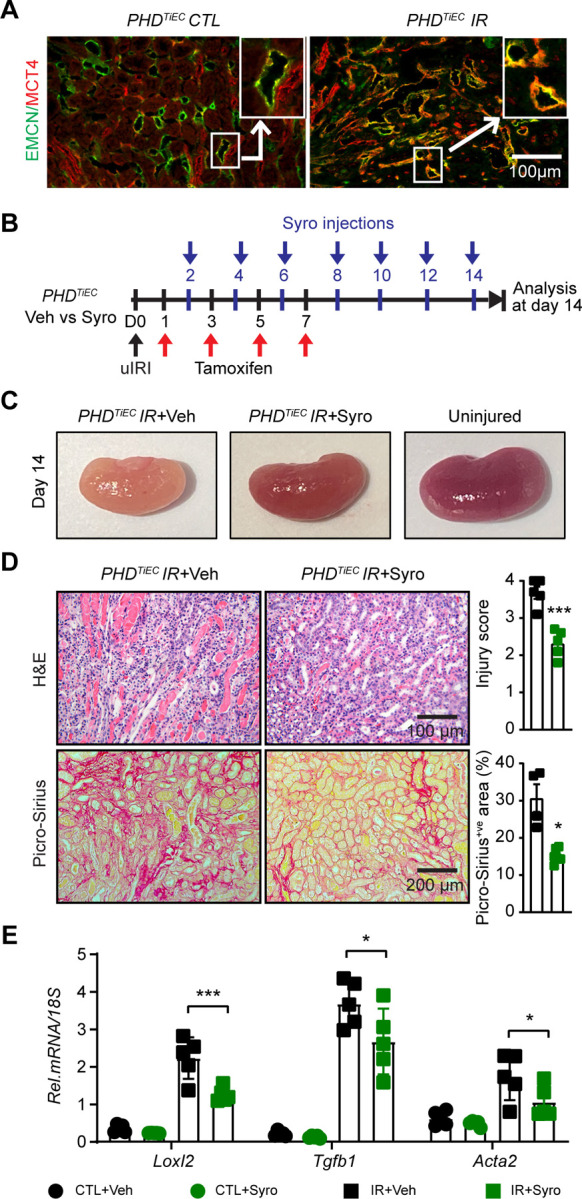
Ischemic acute kidney injury (AKI) is common in hospitalized patients and increases the risk for chronic kidney disease (CKD). Impaired endothelial cell (EC) functions are thought to contribute in AKI to CKD transition, but the underlying mechanisms remain unclear. Here, we identify a critical role for endothelial oxygen sensing prolyl hydroxylase domain (PHD) enzymes 1-3 in regulating post-ischemic kidney repair. In renal endothelium, we observed compartment-specific differences in the expression of the three PHD isoforms in both mice and humans. We found that post-ischemic concurrent inactivation of endothelial PHD1, PHD2, and PHD3 but not PHD2 alone promoted maladaptive kidney repair characterized by exacerbated tissue injury, fibrosis, and inflammation. Single-cell RNA-seq analysis of the post-ischemic endothelial PHD1, PHD2 and PHD3 deficient () kidney revealed an endothelial glycolytic transcriptional signature, also observed in human kidneys with severe AKI. This metabolic program was coupled to upregulation of the gene encoding the lactate exporter monocarboxylate transporter 4 (MCT4). Strikingly, treatment with the MCT4 inhibitor syrosingopine restored adaptive kidney repair in mice. Mechanistically, MCT4 inhibition suppressed pro-inflammatory EC activation reducing monocyte-endothelial cell interaction. Our findings suggest avenues for halting AKI to CKD transition based on selectively targeting the endothelial hypoxia-driven glycolysis/MCT4 axis.
缺血性急性肾损伤(AKI)在住院患者中很常见,并会增加慢性肾脏病(CKD)的风险。内皮细胞(EC)功能受损被认为在AKI向CKD的转变中起作用,但其潜在机制仍不清楚。在此,我们确定了内皮氧感应脯氨酰羟化酶结构域(PHD)酶1-3在调节缺血后肾脏修复中的关键作用。在肾内皮中,我们观察到小鼠和人类中三种PHD异构体表达的区域特异性差异。我们发现,缺血后内皮PHD1、PHD2和PHD3同时失活而非单独失活PHD2会促进适应性不良的肾脏修复,其特征为组织损伤、纤维化和炎症加剧。对缺血后内皮PHD1、PHD2和PHD3缺陷()肾脏的单细胞RNA测序分析揭示了一种内皮糖酵解转录特征,在患有严重AKI的人类肾脏中也观察到了这种特征。这种代谢程序与编码乳酸转运体单羧酸转运体4(MCT4)的基因上调相关。引人注目的是,用MCT4抑制剂西罗辛平治疗可恢复小鼠的适应性肾脏修复。从机制上讲,MCT4抑制可抑制促炎内皮细胞活化,减少单核细胞与内皮细胞的相互作用。我们的研究结果为基于选择性靶向内皮缺氧驱动的糖酵解/MCT4轴来阻止AKI向CKD转变提供了途径。