Jin Liang, Wang Fei, Wang Xue, Harvey Bohdan P, Bi Yingtao, Hu Chenqi, Cui Baoliang, Darcy Anhdao T, Maull John W, Phillips Ben R, Kim Youngjae, Jenkins Gary J, Sornasse Thierry R, Tian Yu
Research & Development, AbbVie, North Chicago, IL 60064, USA.
DMPK, Takeda Development Center Americas Inc., Cambridge, MA 02142, USA.
Proteomes. 2023 Oct 16;11(4):32. doi: 10.3390/proteomes11040032.
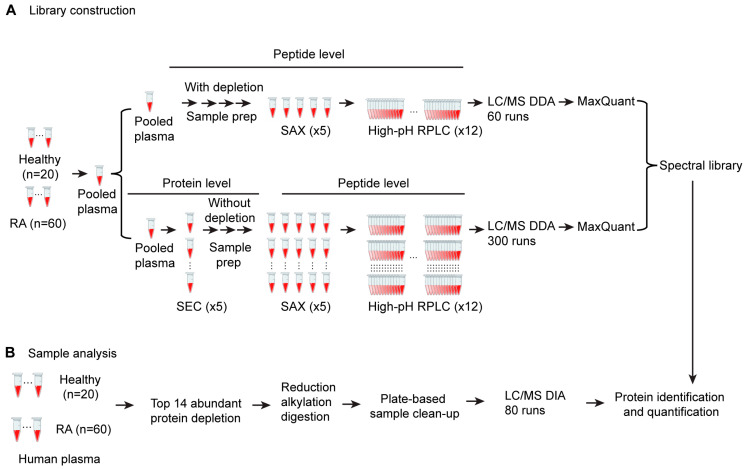
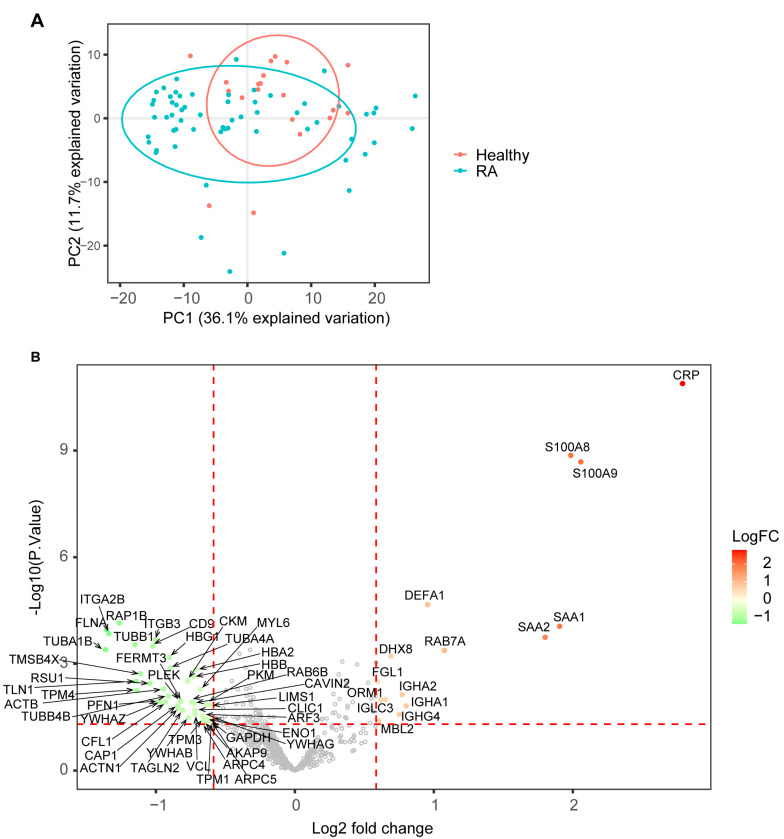
Rheumatoid arthritis (RA) is a systemic autoimmune and inflammatory disease. Plasma biomarkers are critical for understanding disease mechanisms, treatment effects, and diagnosis. Mass spectrometry-based proteomics is a powerful tool for unbiased biomarker discovery. However, plasma proteomics is significantly hampered by signal interference from high-abundance proteins, low overall protein coverage, and high levels of missing data from data-dependent acquisition (DDA). To achieve quantitative proteomics analysis for plasma samples with a balance of throughput, performance, and cost, we developed a workflow incorporating plate-based high abundance protein depletion and sample preparation, comprehensive peptide spectral library building, and data-independent acquisition (DIA) SWATH mass spectrometry-based methodology. In this study, we analyzed plasma samples from both RA patients and healthy donors. The results showed that the new workflow performance exceeded that of the current state-of-the-art depletion-based plasma proteomic platforms in terms of both data quality and proteome coverage. Proteins from biological processes related to the activation of systemic inflammation, suppression of platelet function, and loss of muscle mass were enriched and differentially expressed in RA. Some plasma proteins, particularly acute-phase reactant proteins, showed great power to distinguish between RA patients and healthy donors. Moreover, protein isoforms in the plasma were also analyzed, providing even deeper proteome coverage. This workflow can serve as a basis for further application in discovering plasma biomarkers of other diseases.
类风湿性关节炎(RA)是一种全身性自身免疫性炎症疾病。血浆生物标志物对于理解疾病机制、治疗效果和诊断至关重要。基于质谱的蛋白质组学是一种用于无偏倚生物标志物发现的强大工具。然而,血浆蛋白质组学受到高丰度蛋白质的信号干扰、总体蛋白质覆盖率低以及数据依赖采集(DDA)产生的大量缺失数据的显著阻碍。为了在通量、性能和成本之间取得平衡,实现对血浆样本的定量蛋白质组学分析,我们开发了一种工作流程,该流程结合了基于板的高丰度蛋白质去除和样品制备、综合肽谱库构建以及基于数据非依赖采集(DIA)的SWATH质谱方法。在本研究中,我们分析了RA患者和健康供体的血浆样本。结果表明,在数据质量和蛋白质组覆盖率方面,新的工作流程性能超过了当前基于去除法的最先进血浆蛋白质组学平台。与全身炎症激活、血小板功能抑制和肌肉质量丧失相关的生物学过程中的蛋白质在RA中富集且差异表达。一些血浆蛋白,特别是急性期反应蛋白,在区分RA患者和健康供体方面显示出强大的能力。此外,还分析了血浆中的蛋白质异构体,提供了更深层次的蛋白质组覆盖。该工作流程可为进一步应用于发现其他疾病的血浆生物标志物奠定基础。