Rodríguez-Blanque Raquel, Almazán-Soto Cristina, Piqueras-Sola Beatriz, Sánchez-García Juan Carlos, Reinoso-Cobo Andrés, Menor-Rodríguez María José, Cortés-Martín Jonathan
Research Group CTS1068, Andalusia Research Plan, Junta de Andalucía, 18071 Granada, Spain.
San Cecilio University Hospital, 18071 Granada, Spain.
J Clin Med. 2023 Oct 23;12(20):6694. doi: 10.3390/jcm12206694.
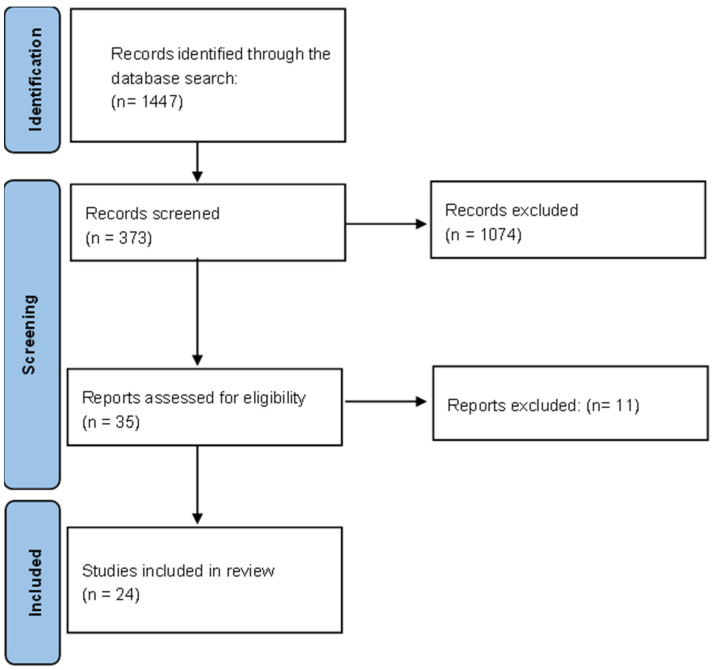
Arnold Chiari syndrome is a rare congenital disease of unknown prevalence and whose origin is still under study. It is encompassed within the posterior cranial malformations, showing a wide spectrum of symptomatology that can range from severe headache, dizziness, and paresthesia to complete asymptomatology. It is for this reason that early diagnosis of the disease is difficult, and it is usually diagnosed in adolescence. Treatment is based on remodeling and decompression of the malformed posterior cranial fossa, although the risk of residual symptoms after surgery is high. The aim of this review is to update all the existing information on this pathology by means of an exhaustive analysis covering all the scientific literature produced in the last 5 years. In addition, it has been carried out following the PRISMA model and registered in PROSPERO with code CRD42023394490. One of the main conclusions based on the results obtained in this review is that the origin of the syndrome could have a genetic basis and that the treatment of choice is the decompression of the posterior cerebral fossa.
阿诺德-奇阿里综合征是一种罕见的先天性疾病,其患病率未知,病因仍在研究中。它属于后颅窝畸形,症状表现范围广泛,从严重头痛、头晕和感觉异常到完全无症状。正因如此,该病早期诊断困难,通常在青少年期被诊断出来。治疗基于对畸形后颅窝进行重塑和减压,尽管手术后残留症状的风险很高。本综述的目的是通过对过去5年发表的所有科学文献进行详尽分析,更新关于这种病理状况的所有现有信息。此外,本综述遵循PRISMA模型进行,并已在PROSPERO中注册,注册号为CRD42023394490。基于本综述所得结果的主要结论之一是,该综合征的病因可能有遗传基础,而首选治疗方法是后颅窝减压。